

Pharmacotherapy for pulmonary arterial hypertension
- PMID: 31632754
- PMCID: PMC6783726
- DOI: 10.21037/jtd.2019.09.14
Pharmacotherapy for pulmonary arterial hypertension
Abstract
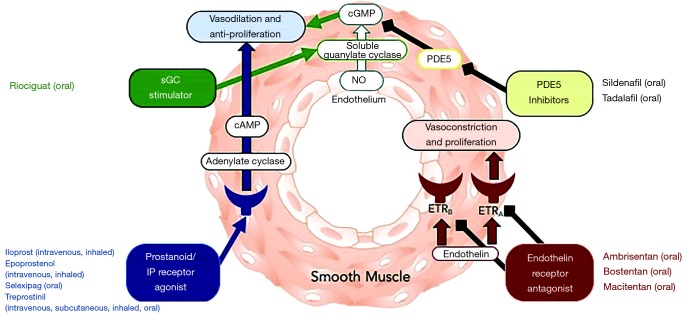
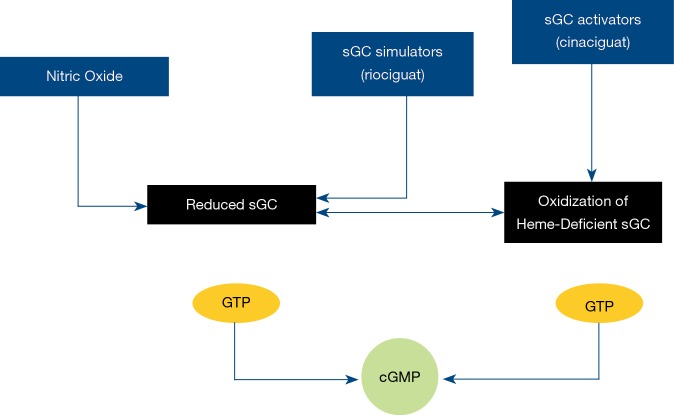
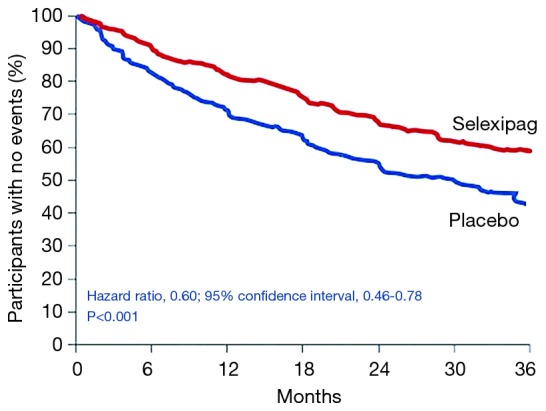
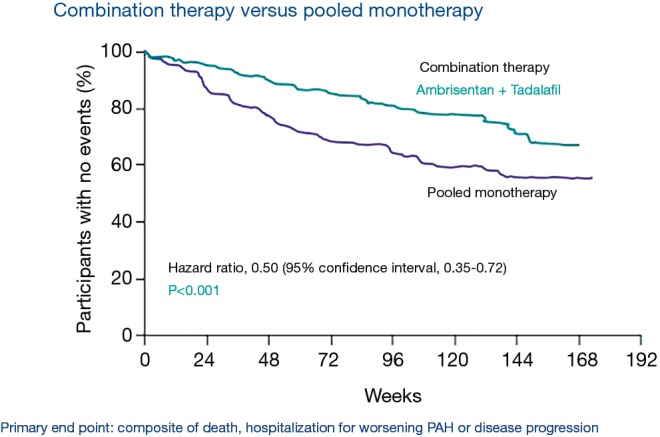
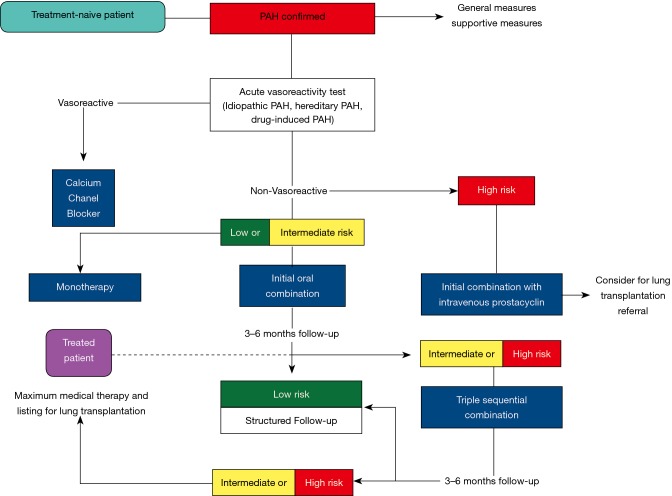
Pulmonary arterial hypertension (PAH) is a condition associated with substantial morbidity and mortality. Over the last 25 years there has been a significant evolution in the therapies to treat PAH. These therapies are effective for patients with group I PAH and group IV PH [chronic thromboembolic pulmonary hypertension (CTEPH)]. PAH is characterized by an imbalance of nitric oxide, prostacyclin and endothelin levels, and current pharmacotherapy involves these three pathways. Earlier clinical trials involving PAH-specific therapies evaluated improvements in 6-minute walk time as a primary improvement whereas contemporary trials have been larger and focused on morbidity and mortality reductions. While there may be a role for monotherapy in disease management, most patients should be considered for dual or triple therapy.
Keywords: Pulmonary arterial hypertension (PAH); ambrisentan; bosentan; endothelin; epoprostenol; macitentan; nitric oxide; prostacyclin; riociguat; selexipag; sildenafil; tadalafil; treprostinil.
2019 Journal of Thoracic Disease. All rights reserved.
Conflict of interest statement
Conflicts of Interest: The authors have no conflicts of interest to declare.
Figures
References
-
- Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015;46:903-75. 10.1183/13993003.01032-2015 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources