

Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition)
- PMID: 31633216
- PMCID: PMC7350392
- DOI: 10.1002/eji.201970107
Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition)
Abstract
These guidelines are a consensus work of a considerable number of members of the immunology and flow cytometry community. They provide the theory and key practical aspects of flow cytometry enabling immunologists to avoid the common errors that often undermine immunological data. Notably, there are comprehensive sections of all major immune cell types with helpful Tables detailing phenotypes in murine and human cells. The latest flow cytometry techniques and applications are also described, featuring examples of the data that can be generated and, importantly, how the data can be analysed. Furthermore, there are sections detailing tips, tricks and pitfalls to avoid, all written and peer-reviewed by leading experts in the field, making this an essential research companion.
© 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Conflict of interest statement
Conflict of interest
Some of the authors of these guidelines work for companies manufacturing FCM equipment and reagents. The mention of a particular company's equipment or reagents does not imply endorsement of these products but are included as examples. The content of these guidelines is editorially independent of any company and has been peer-reviewed.
Figures


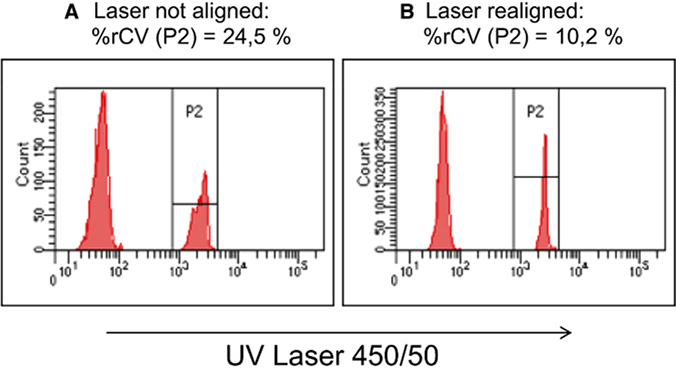
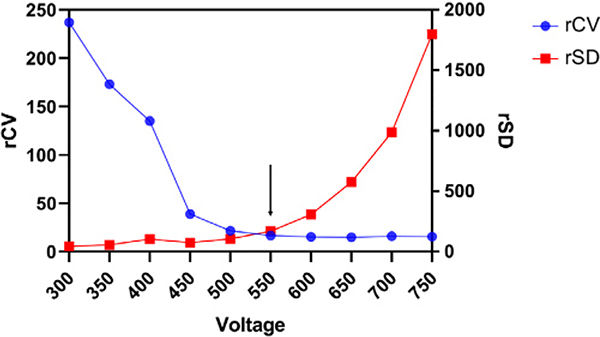
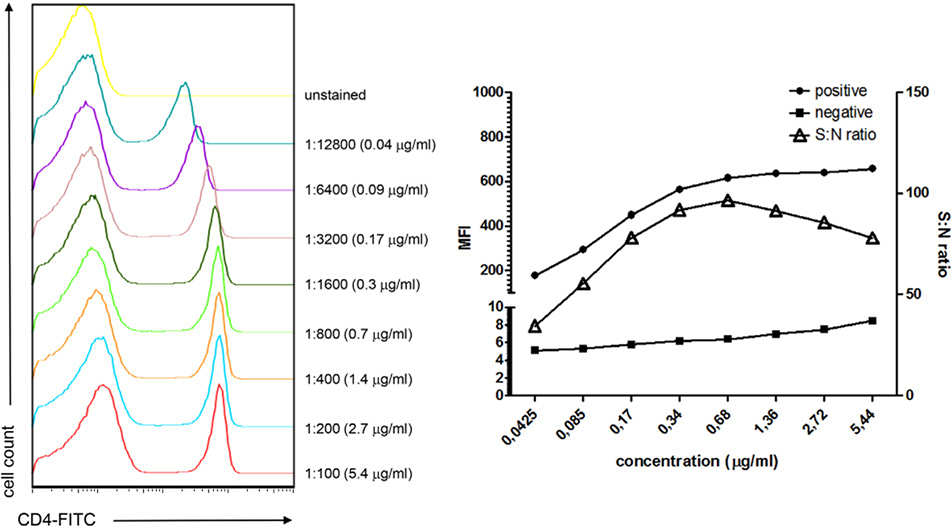
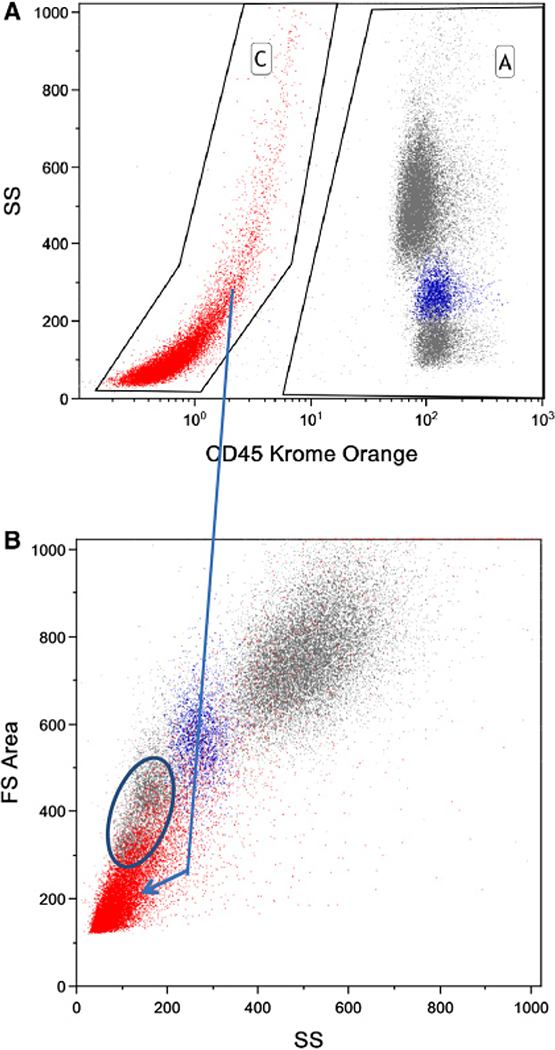



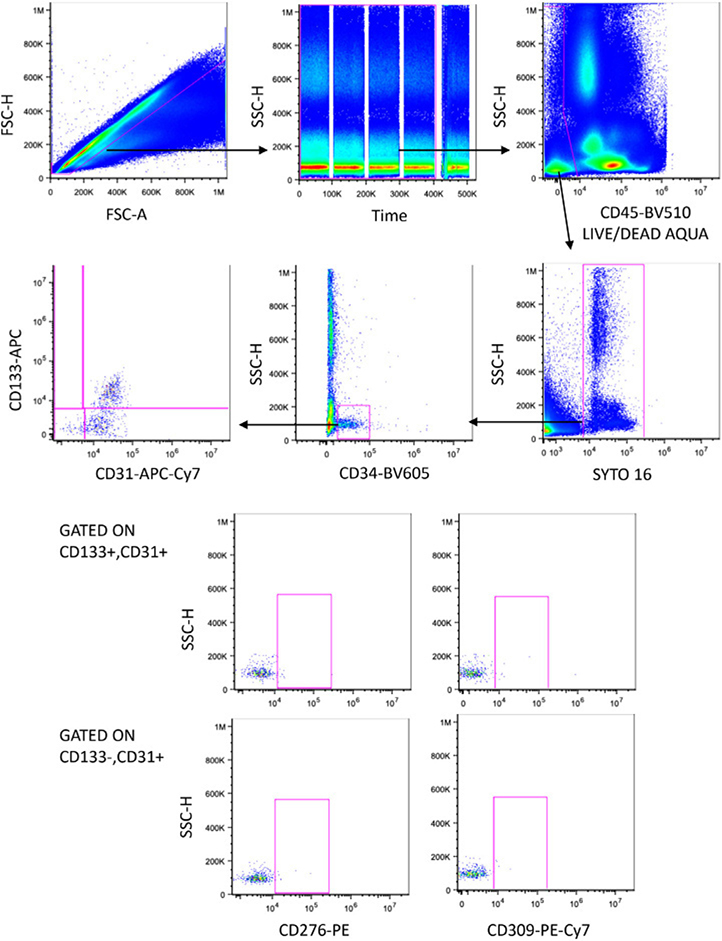

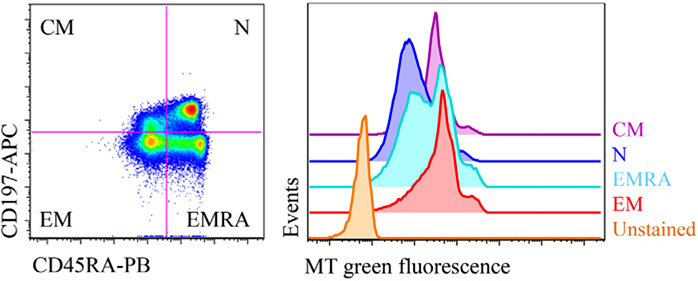
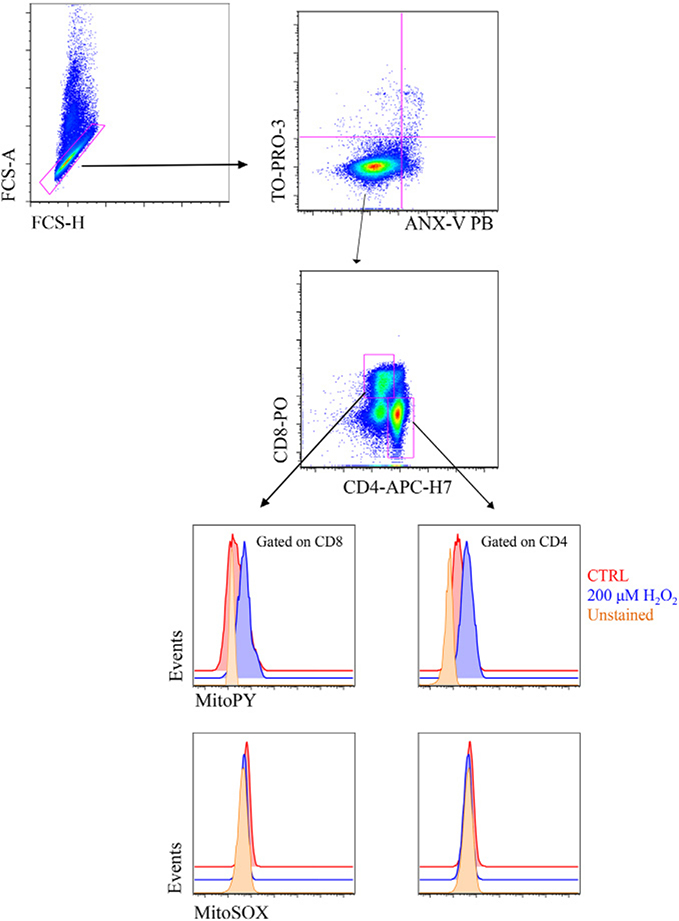
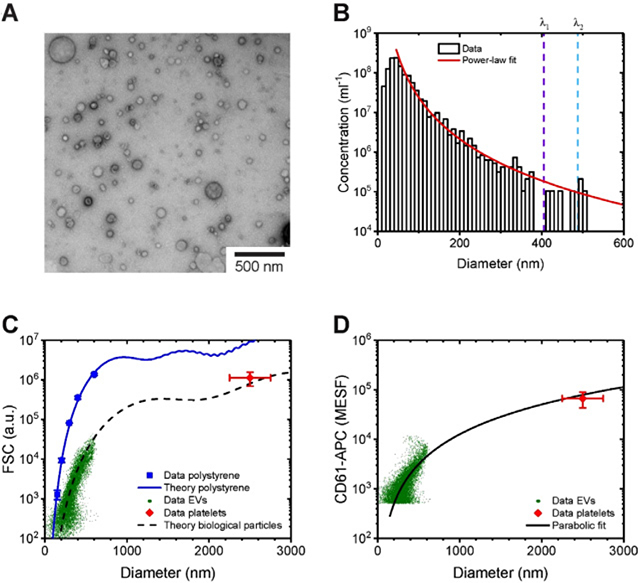



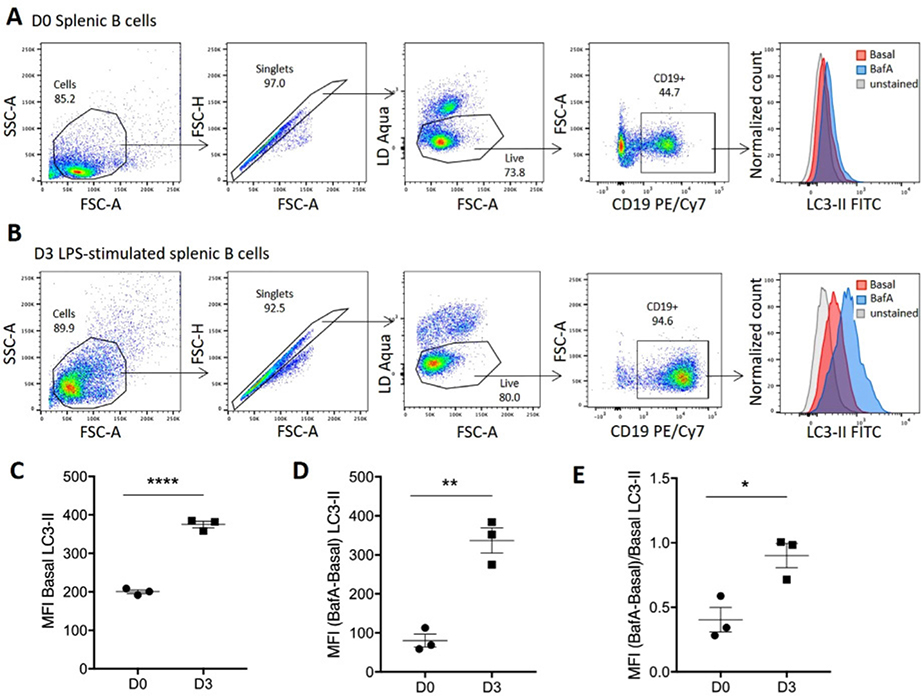
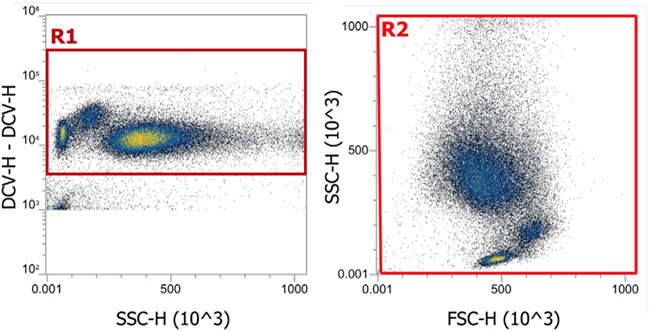
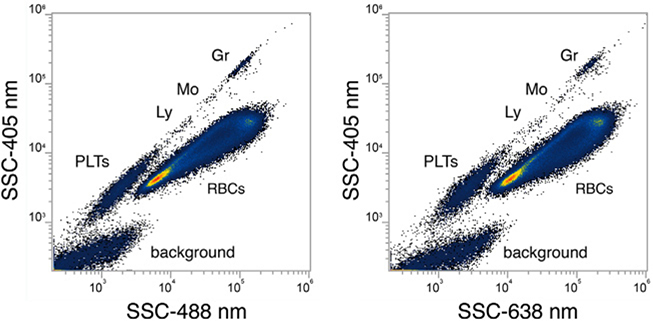
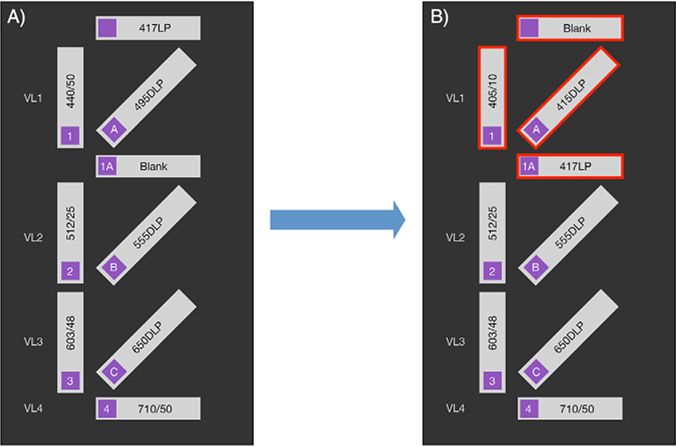

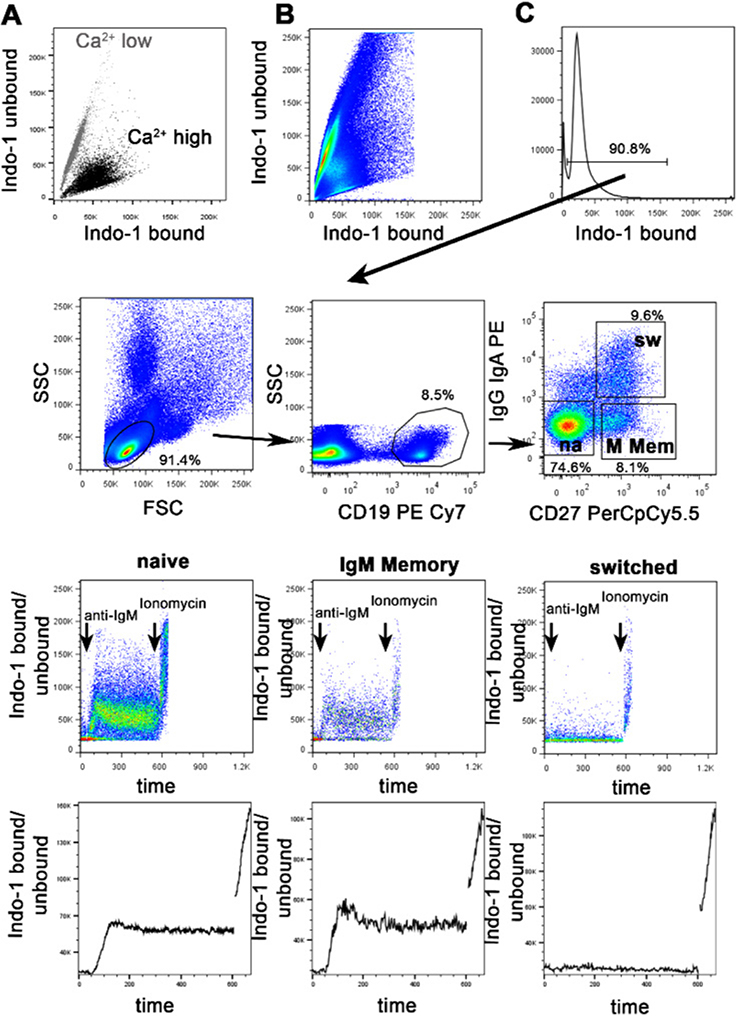
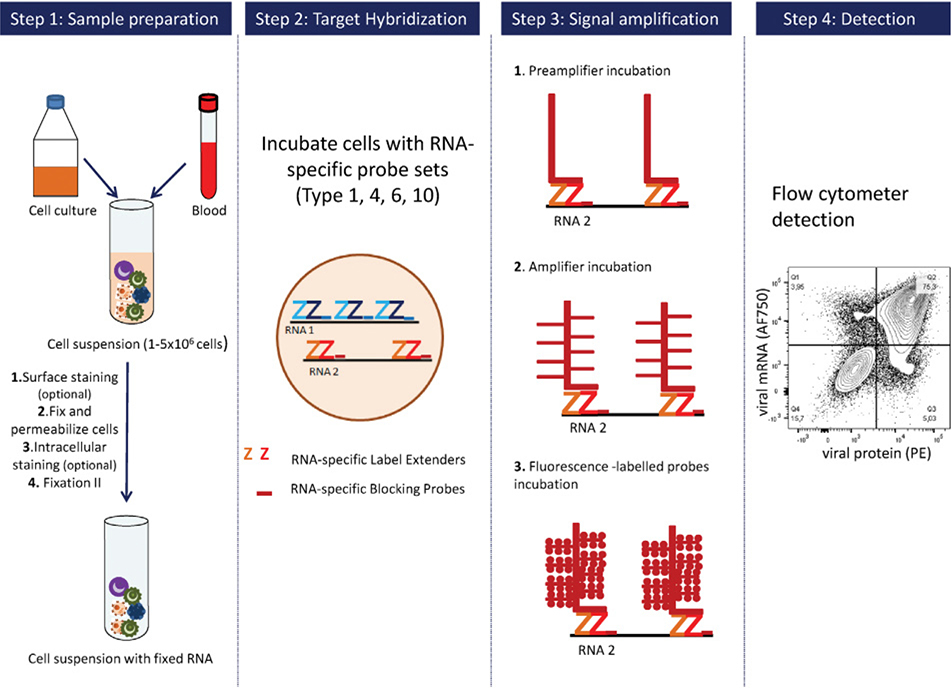
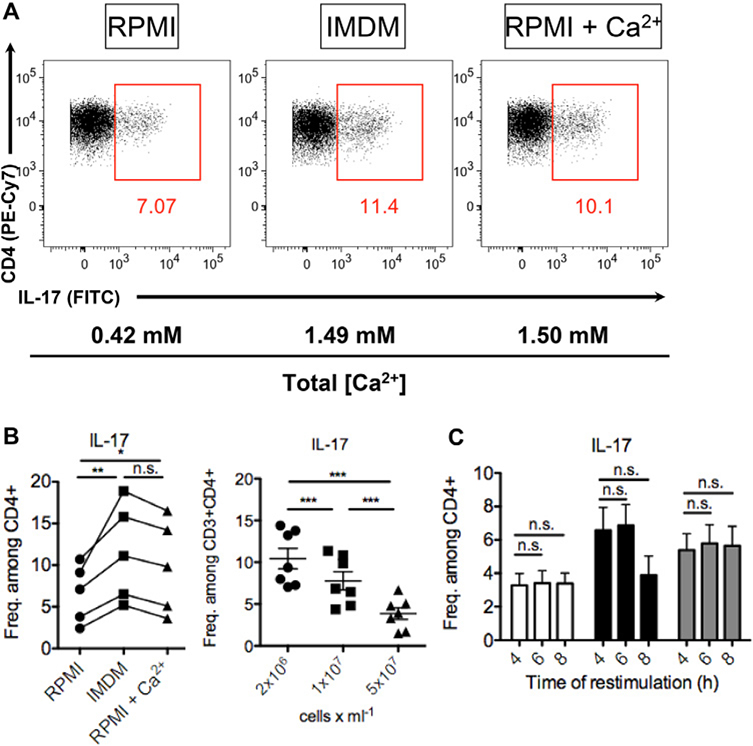
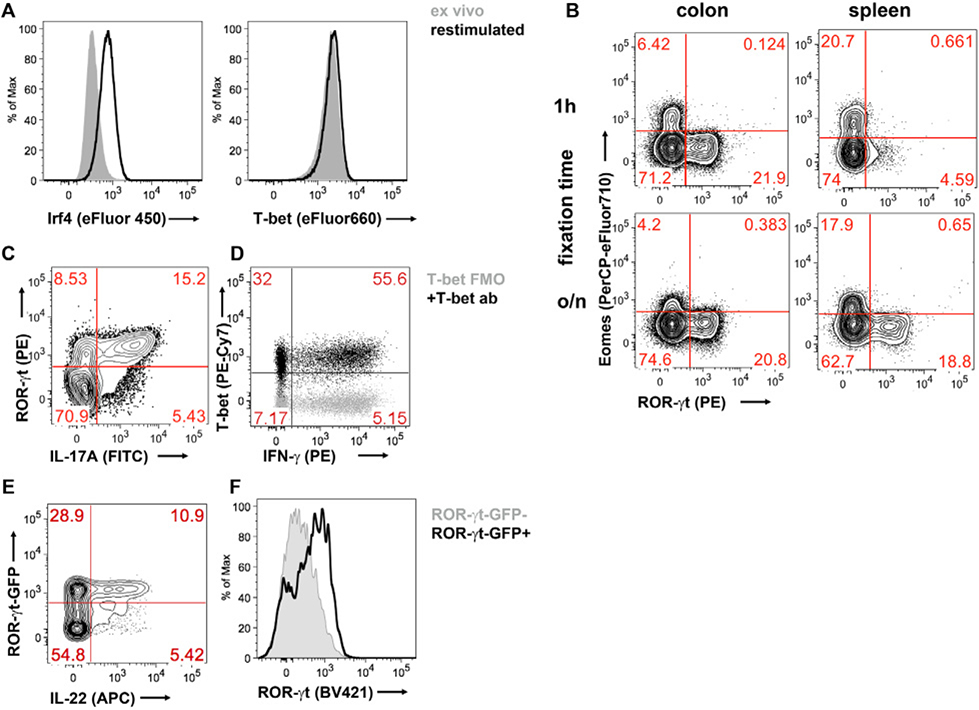


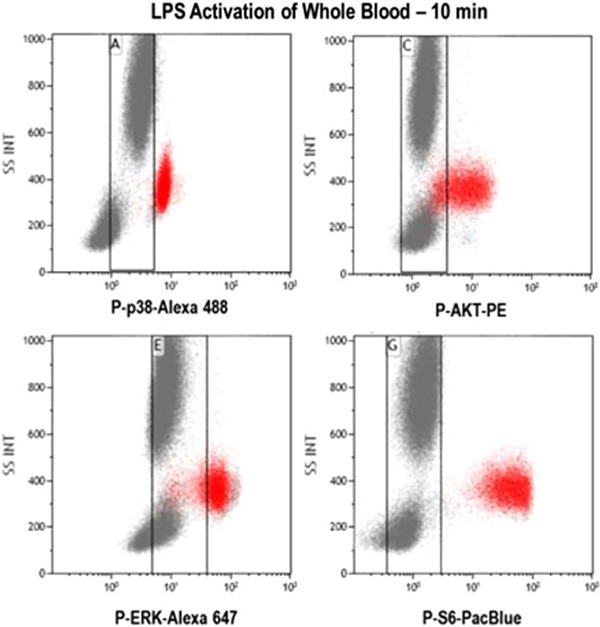
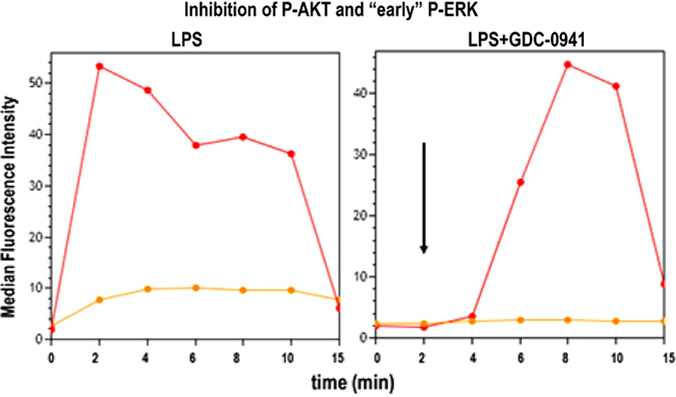
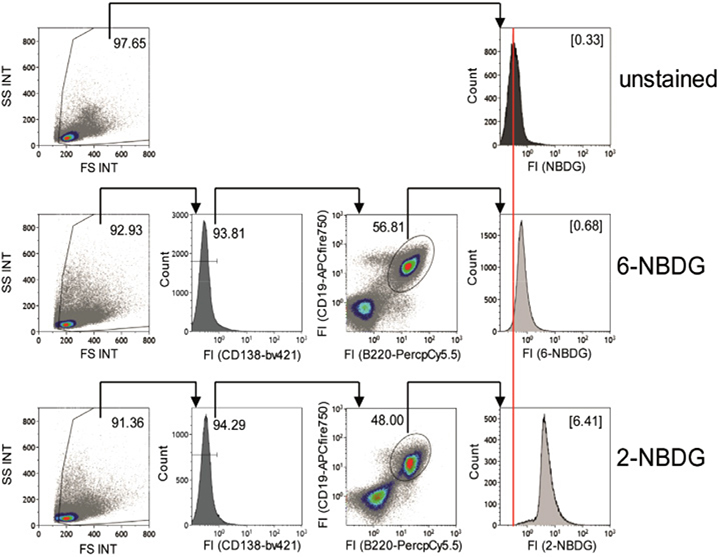
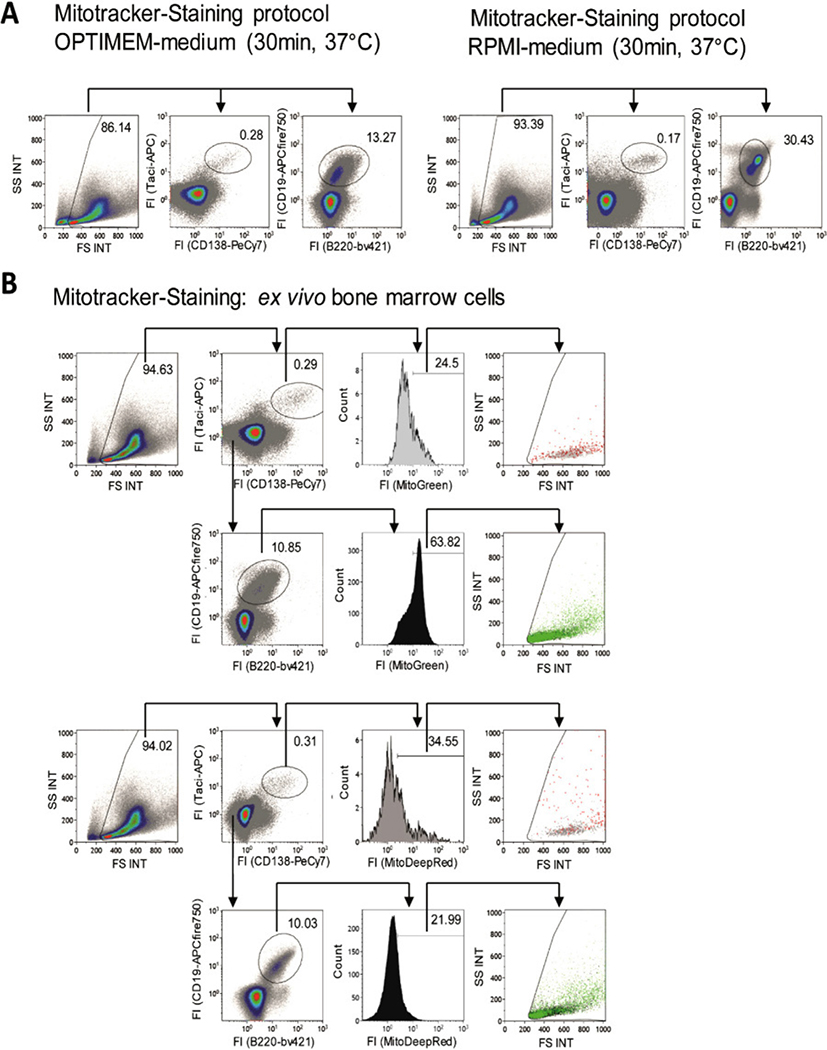


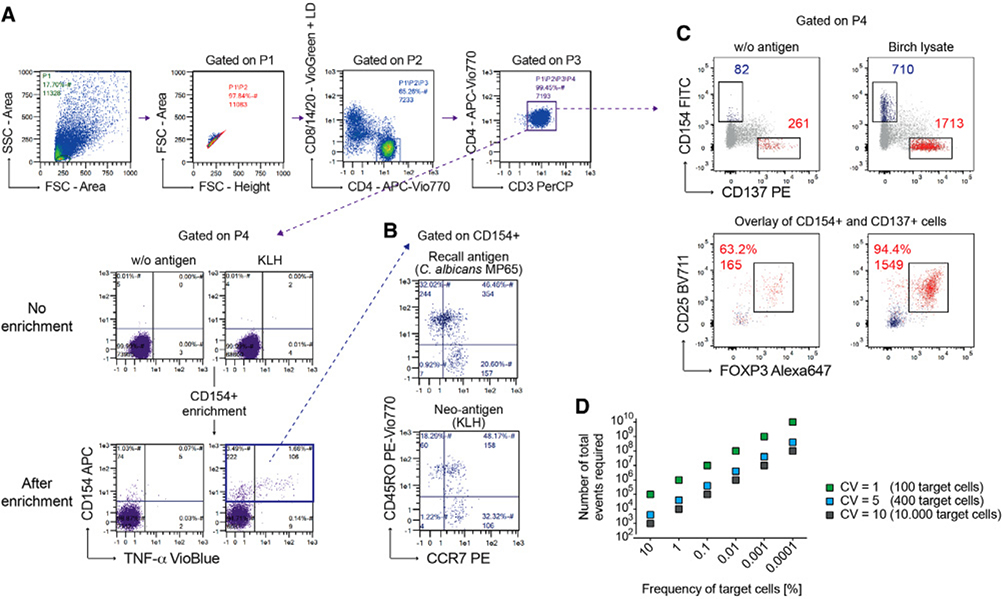
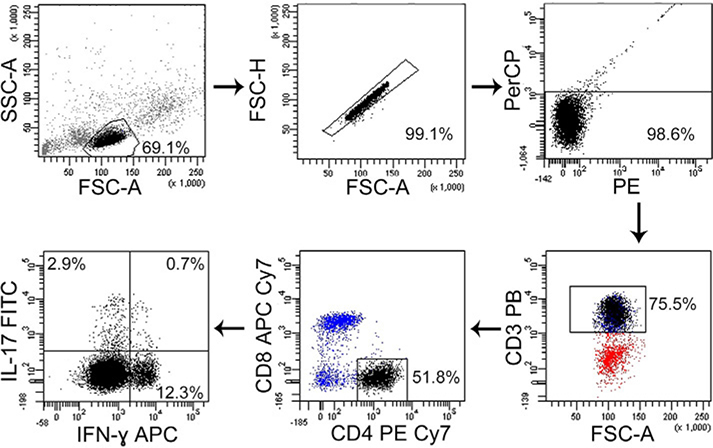
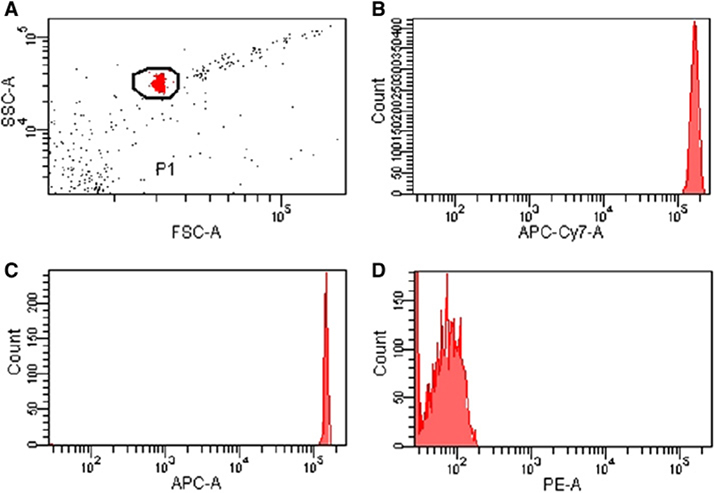
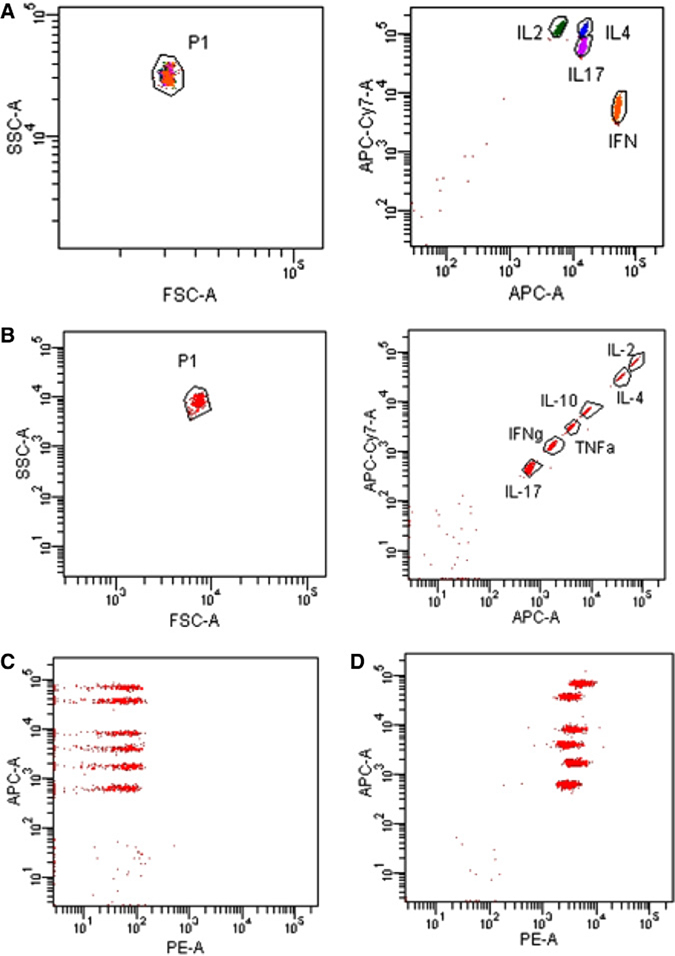
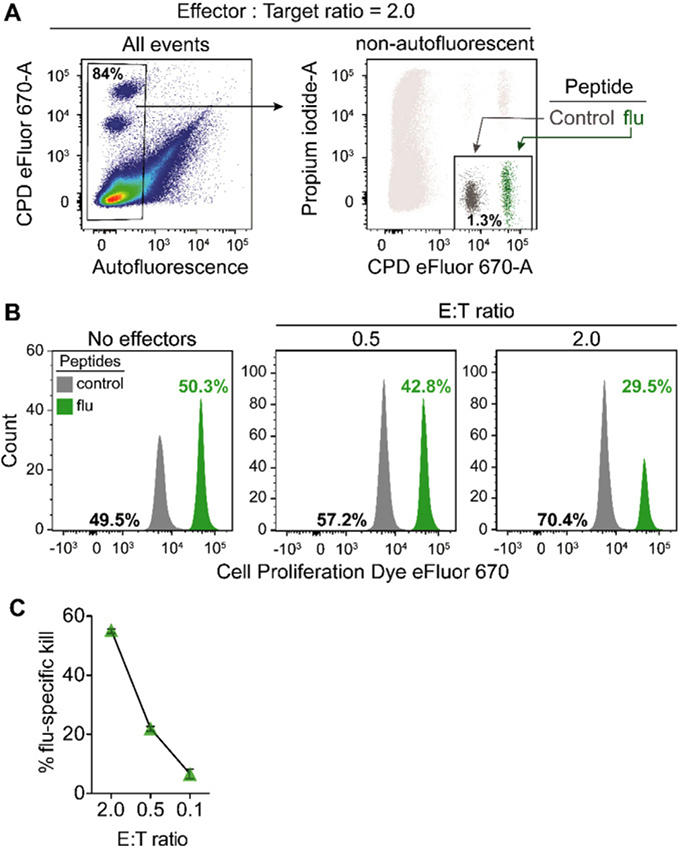
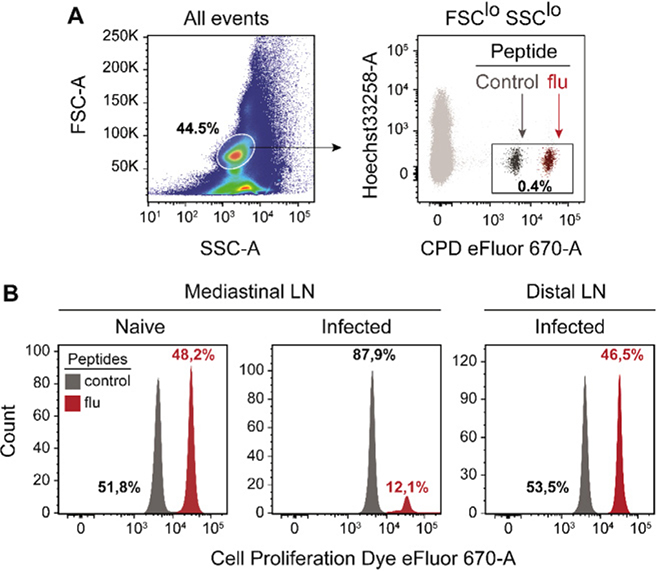


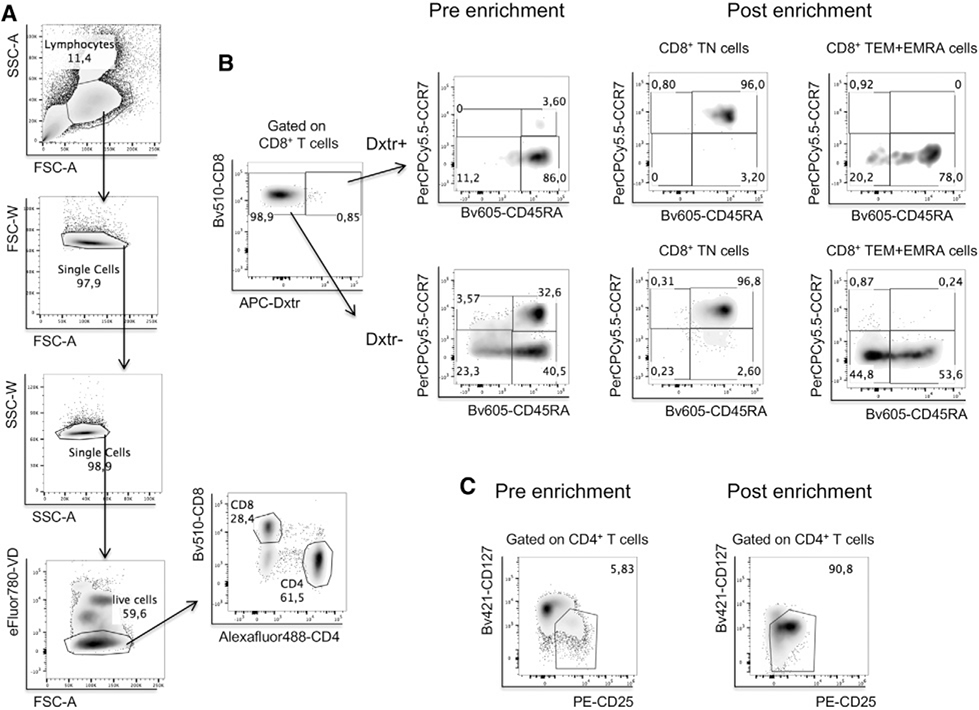
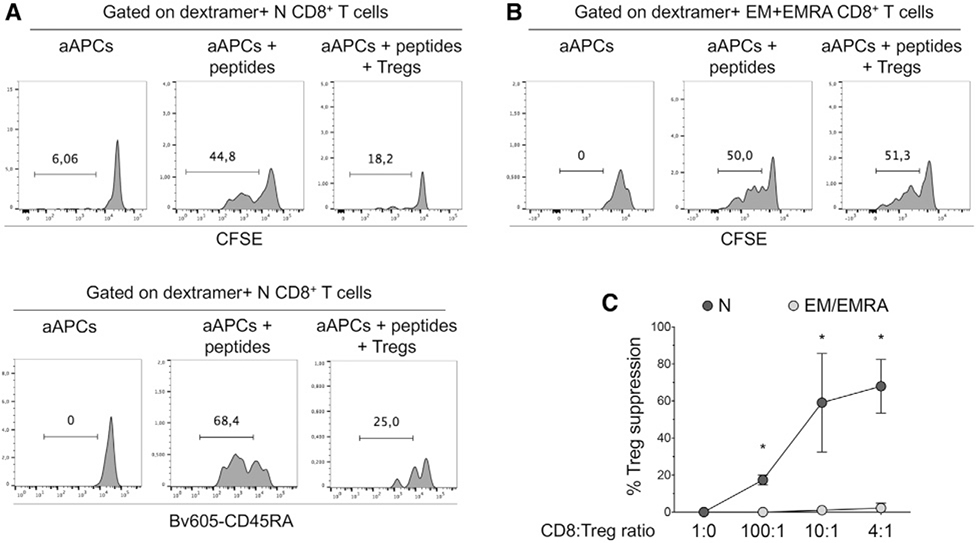
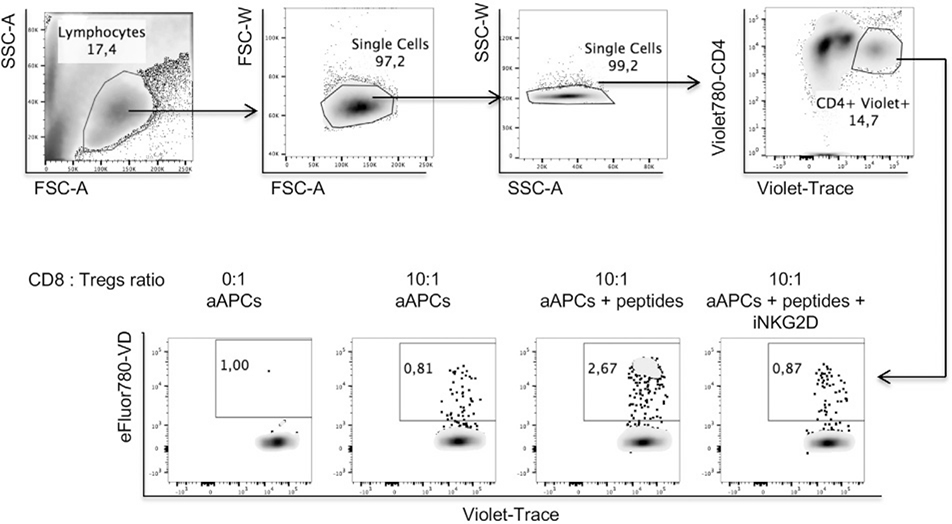
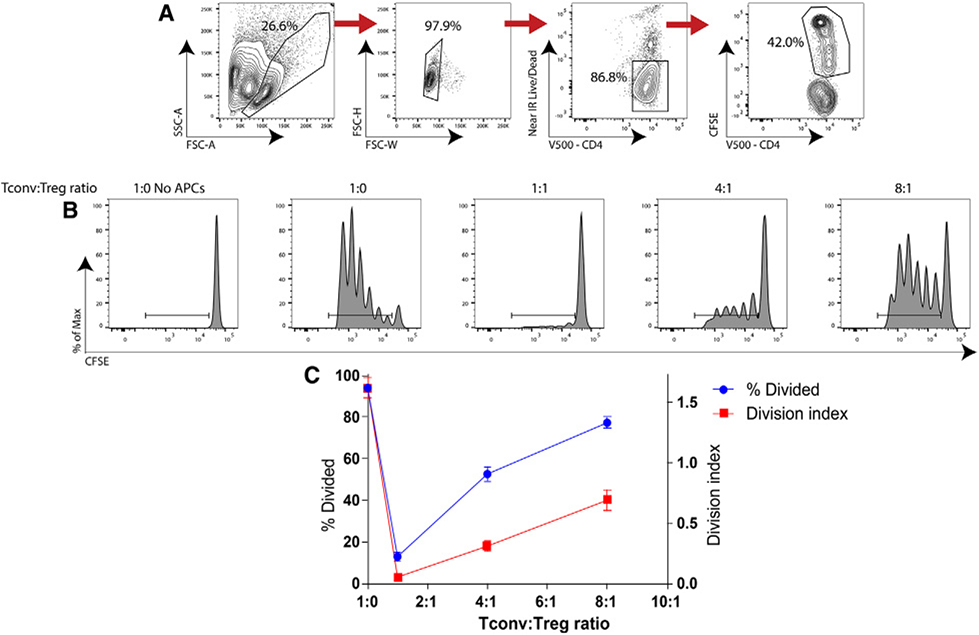
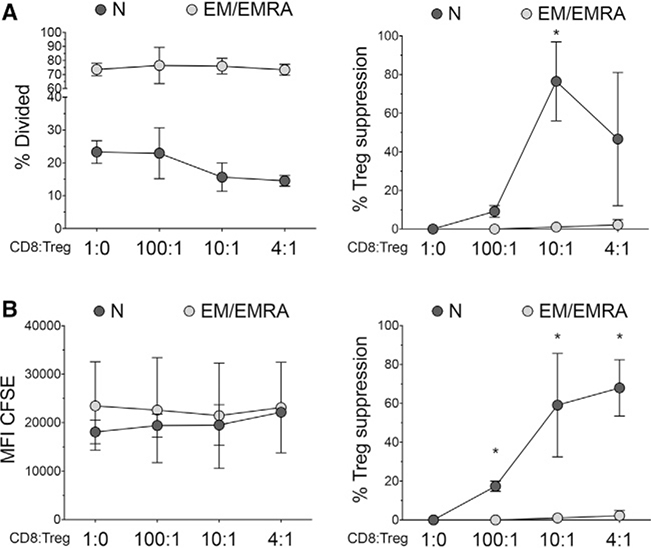
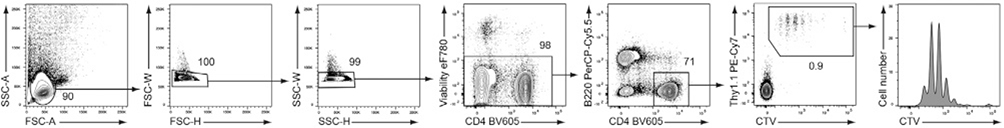
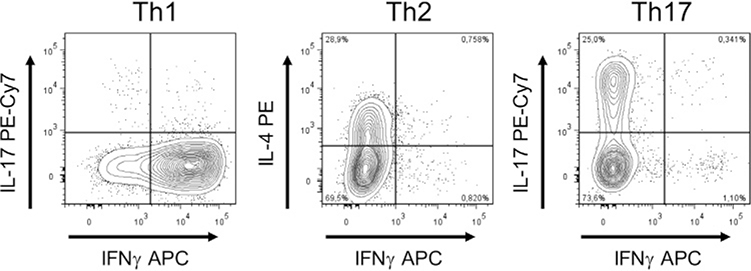
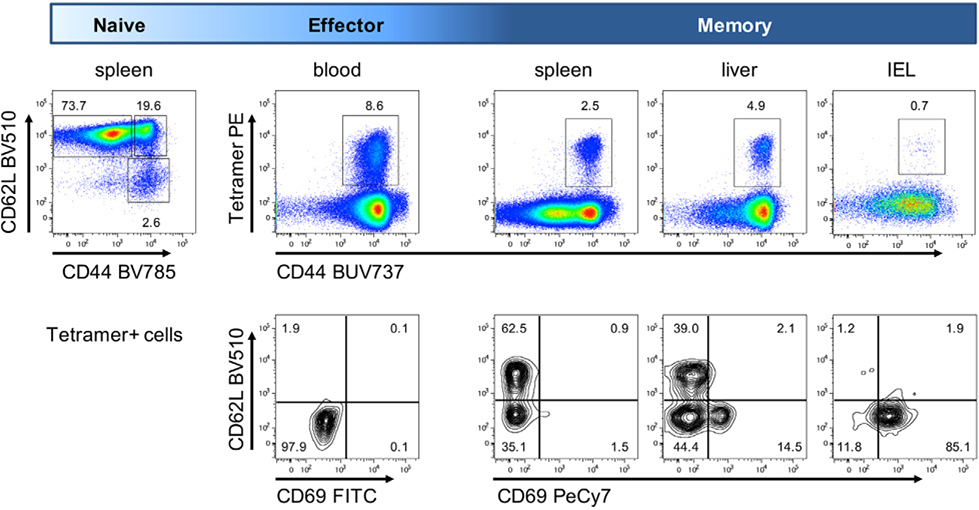

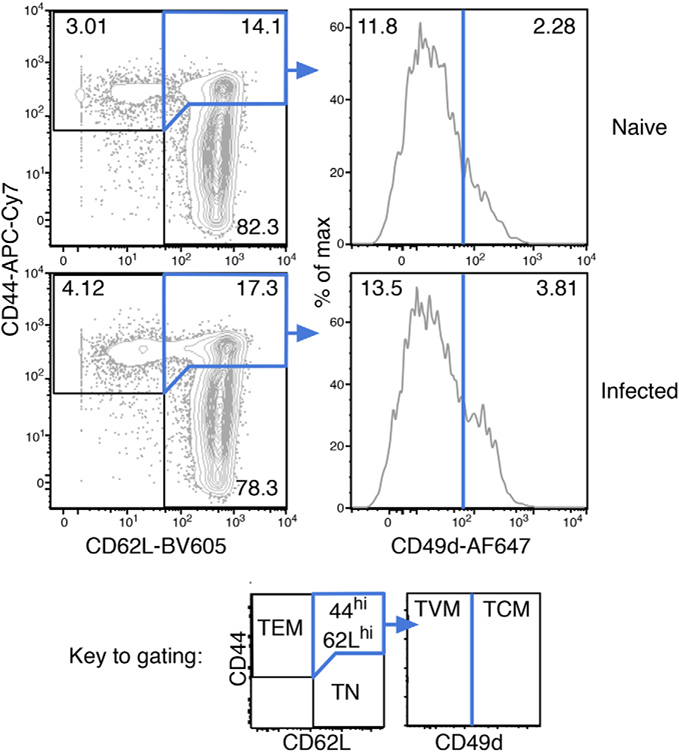
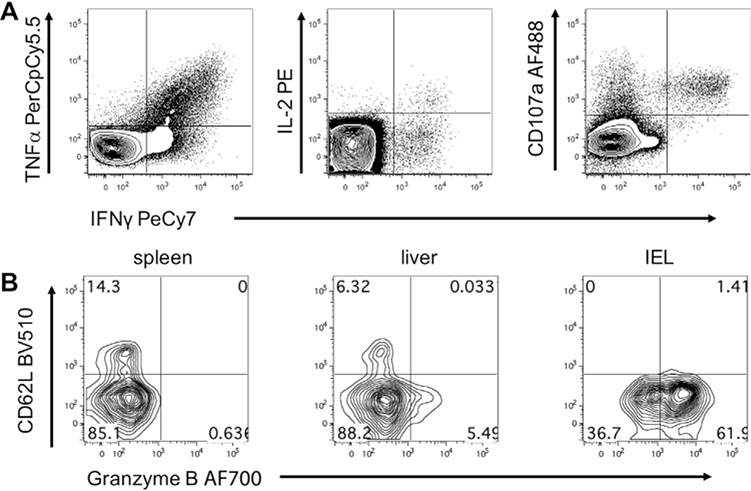

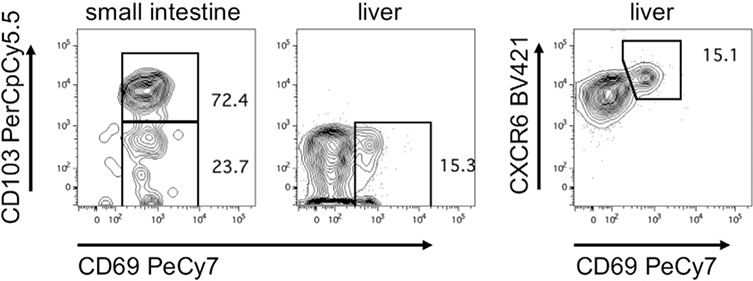
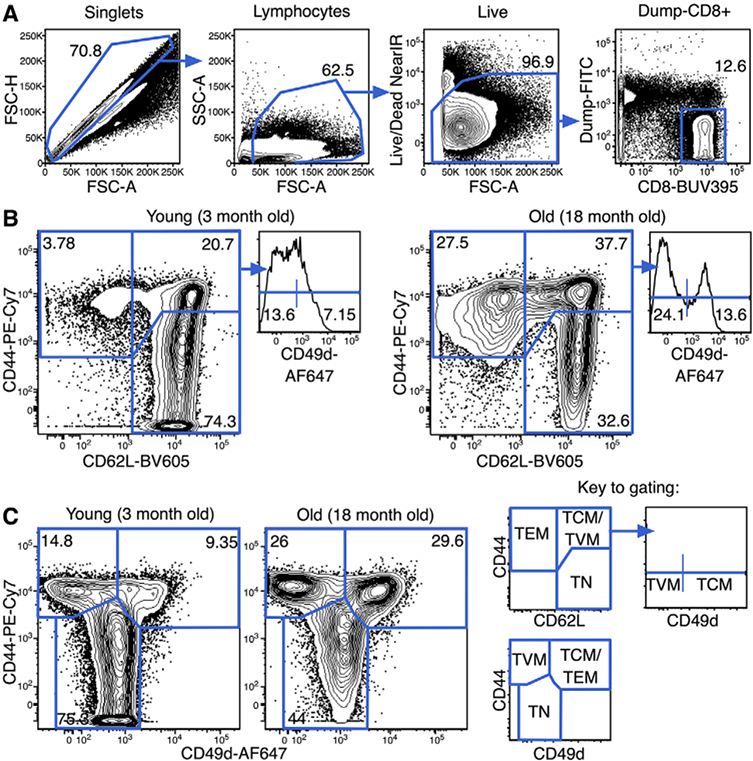
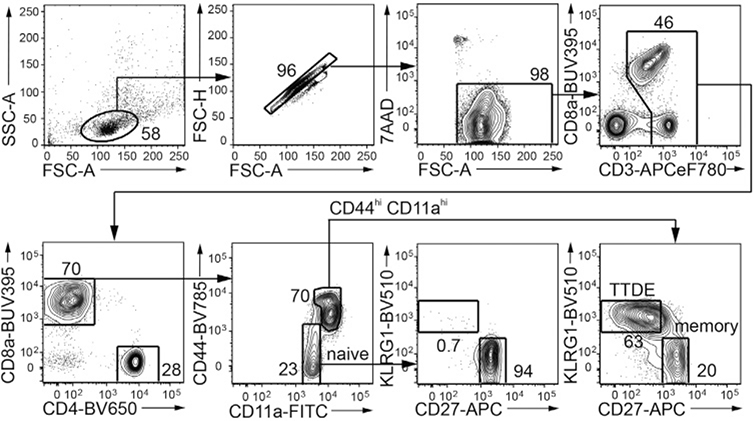
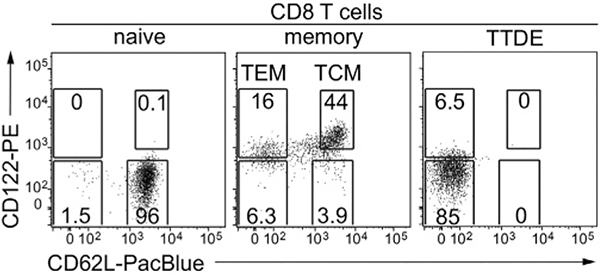
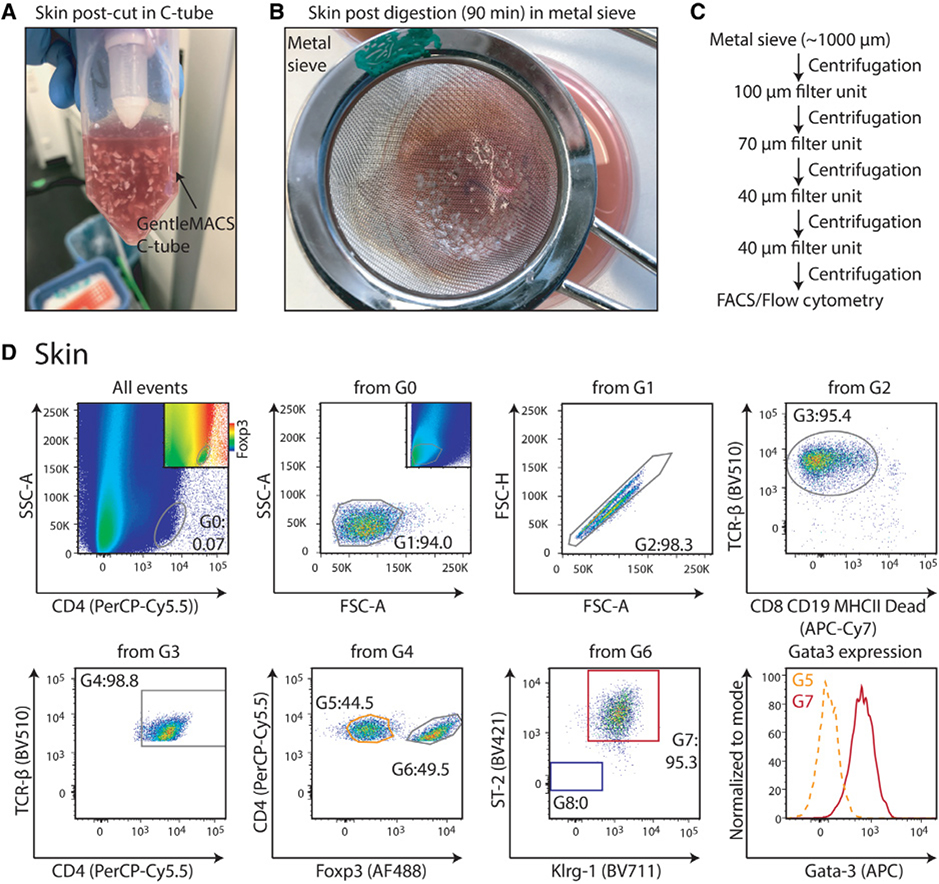
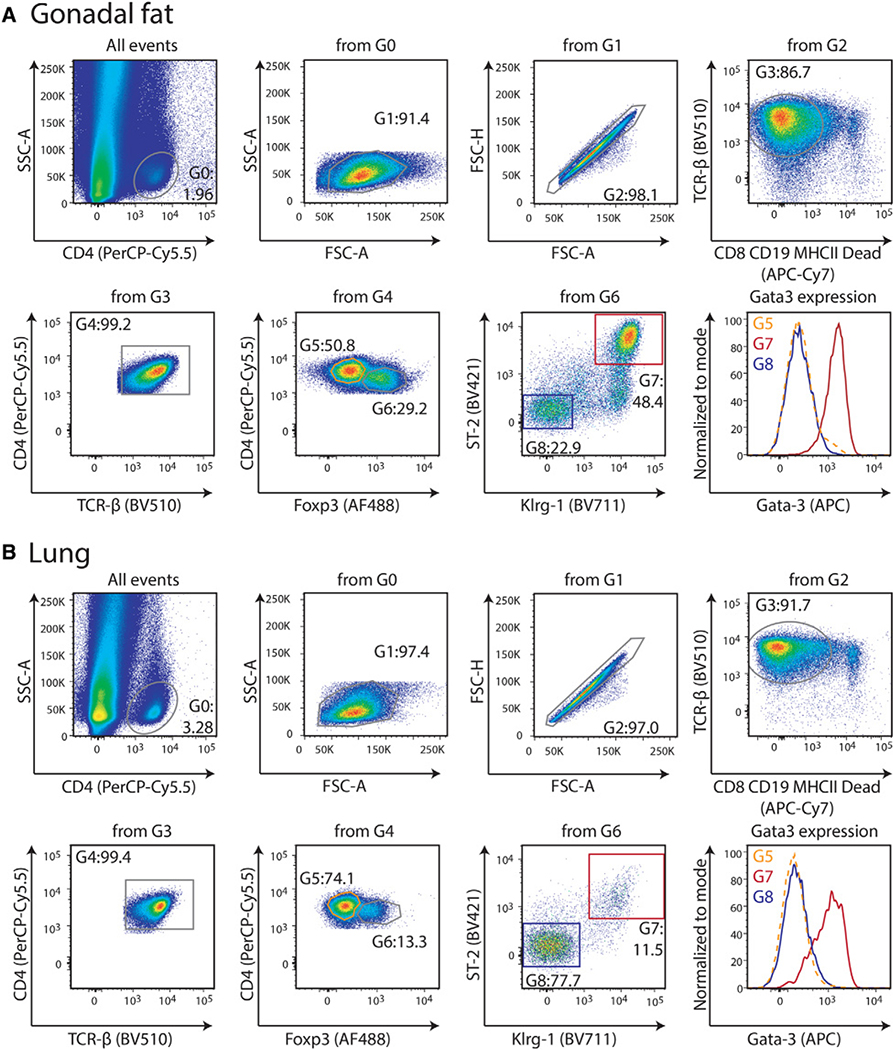
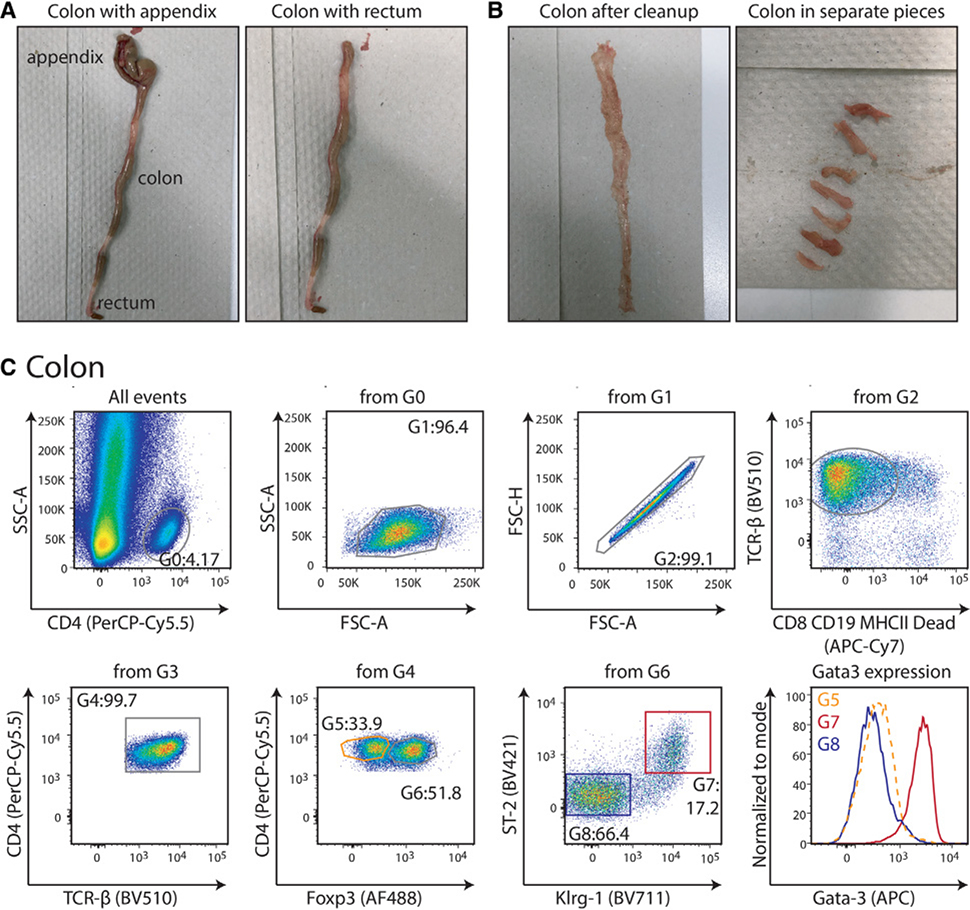
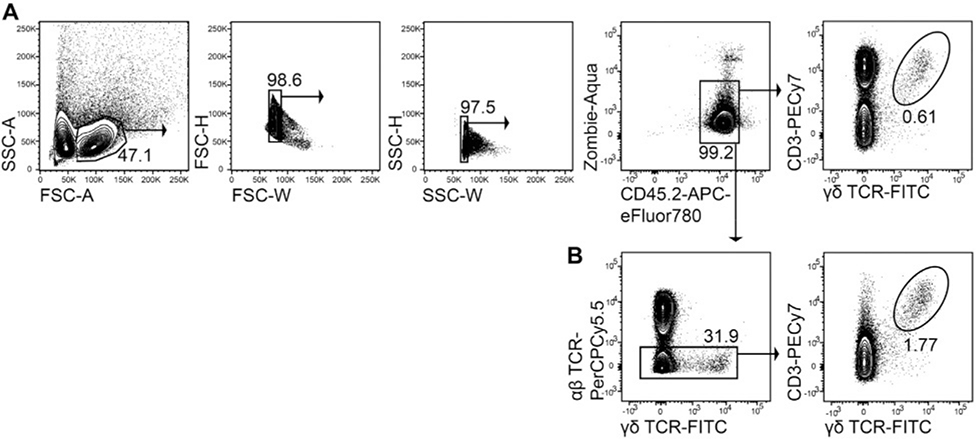
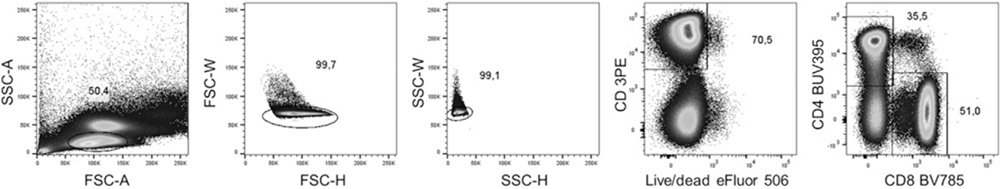
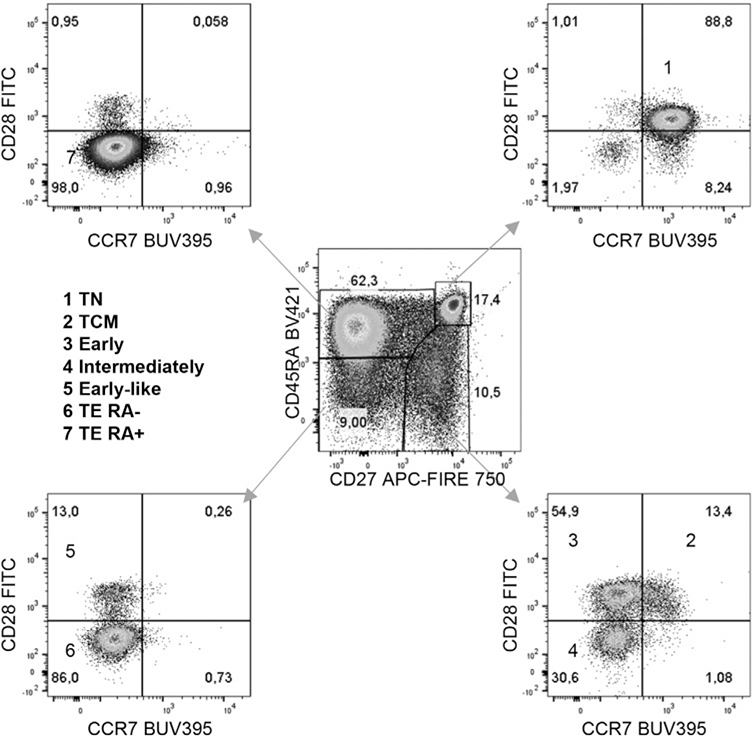
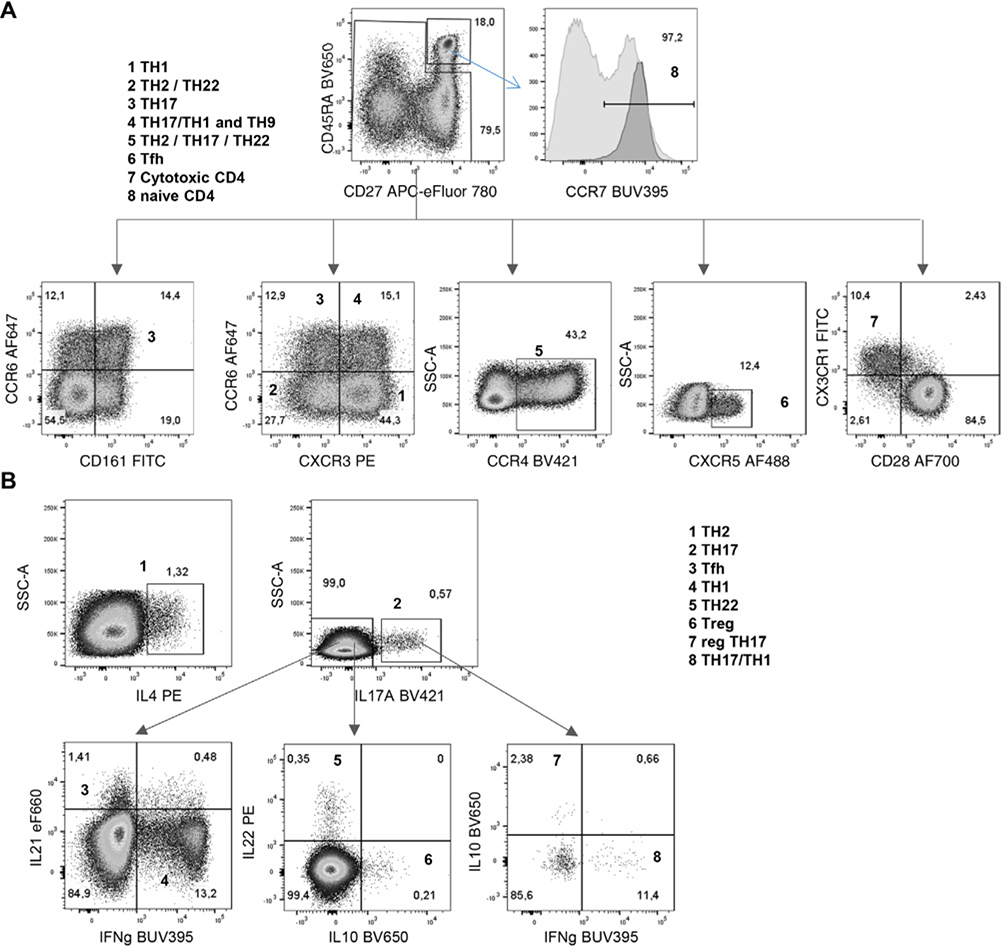
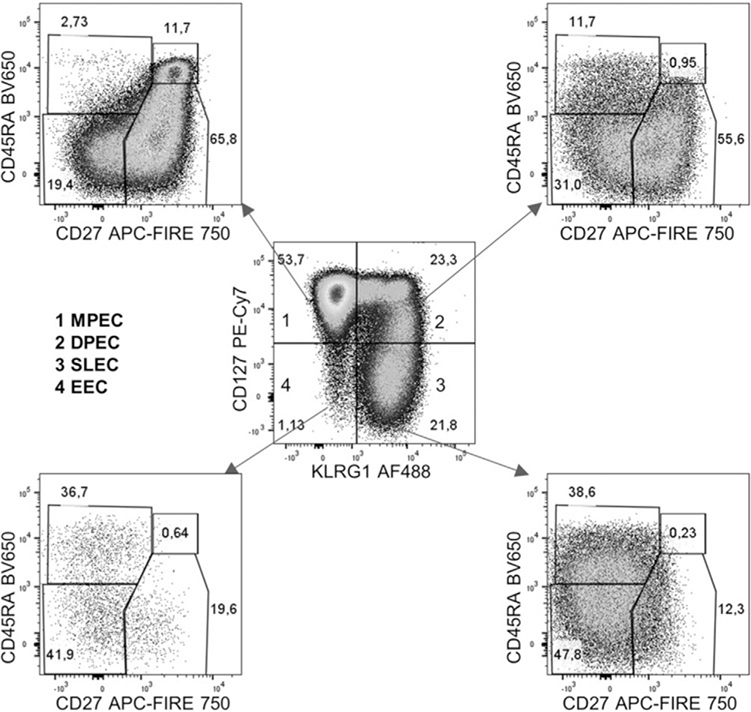
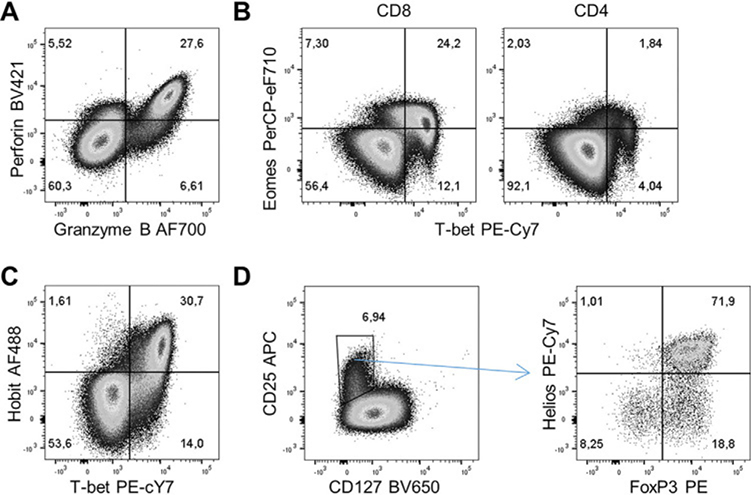
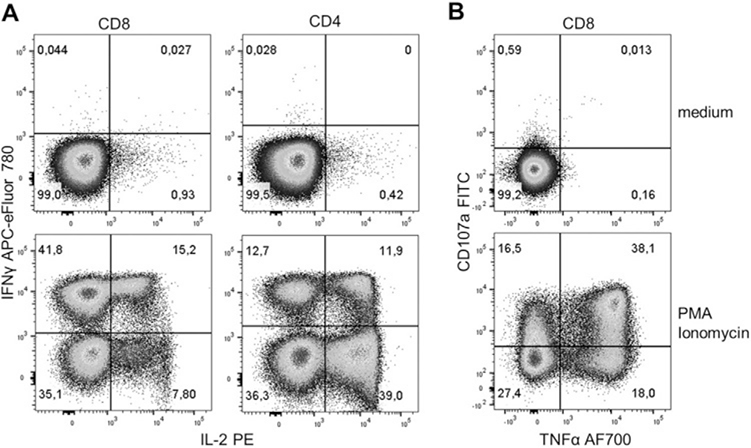
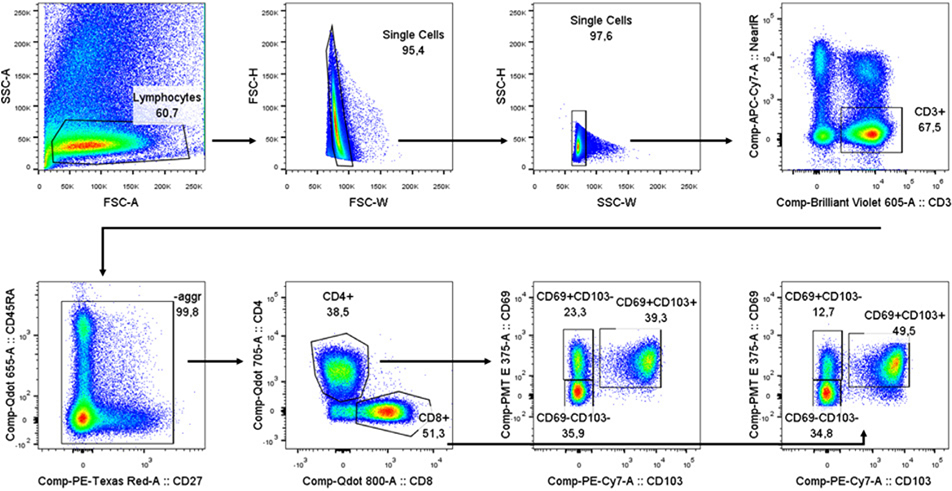
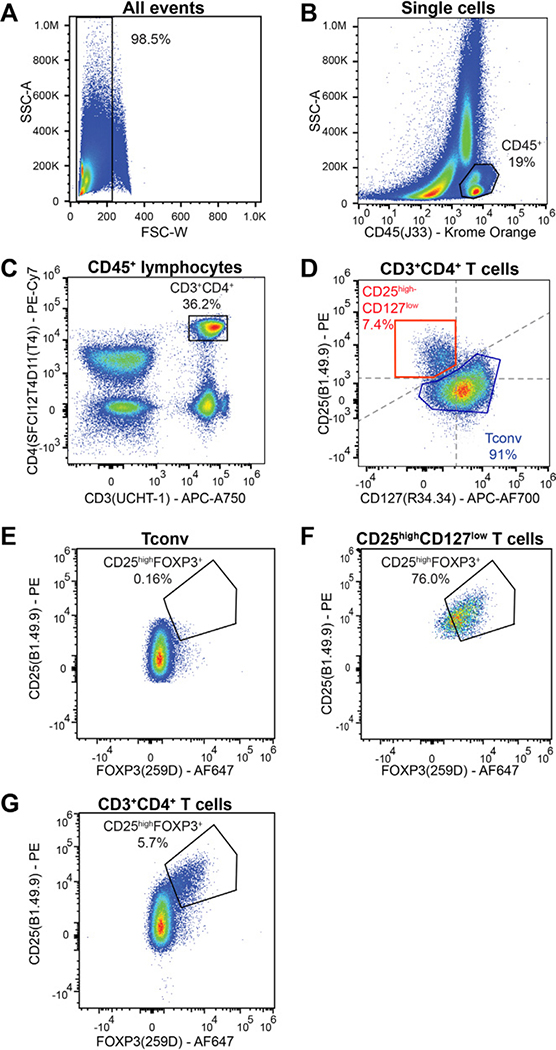
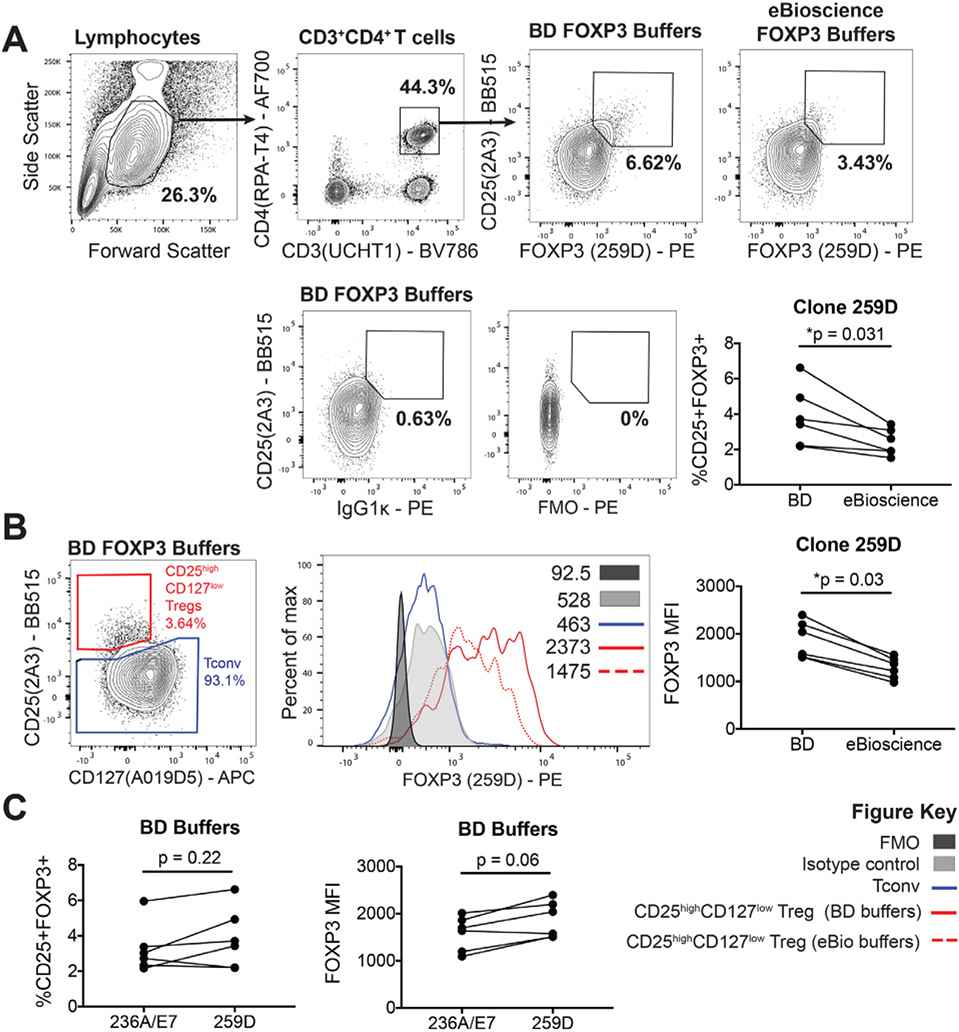
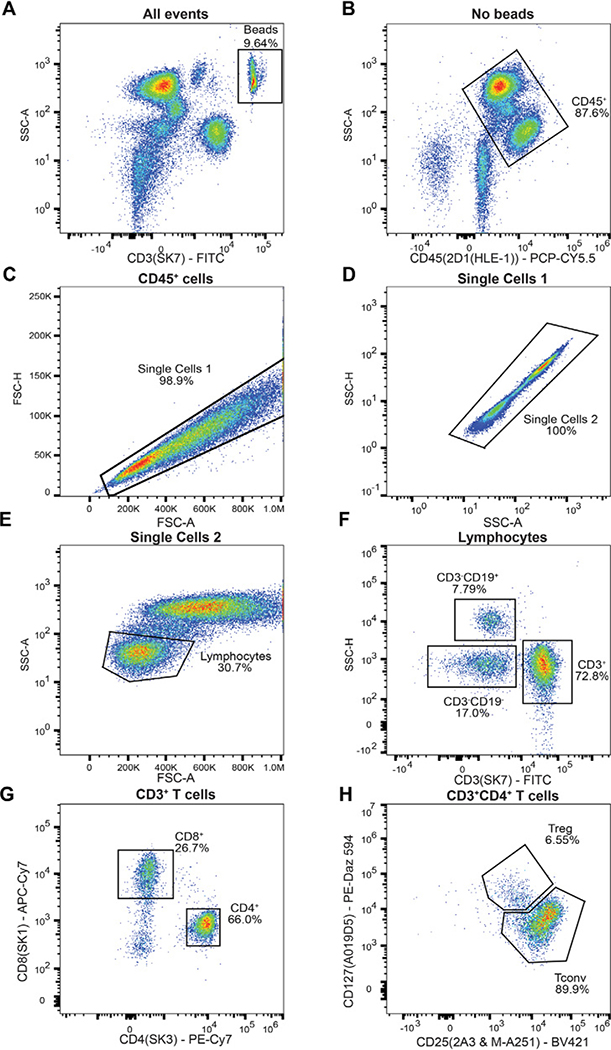
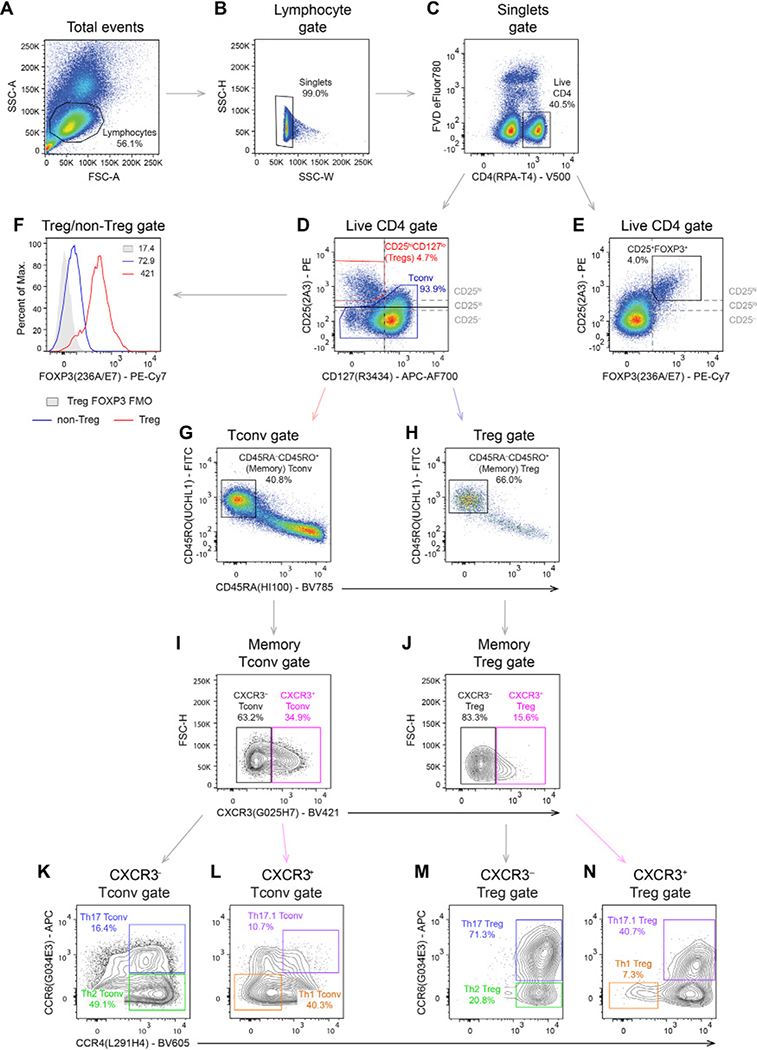

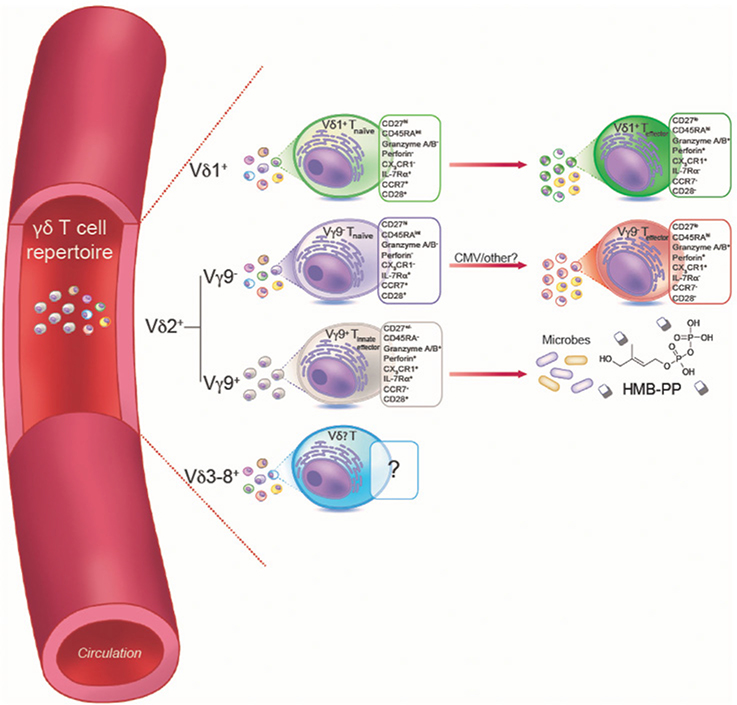
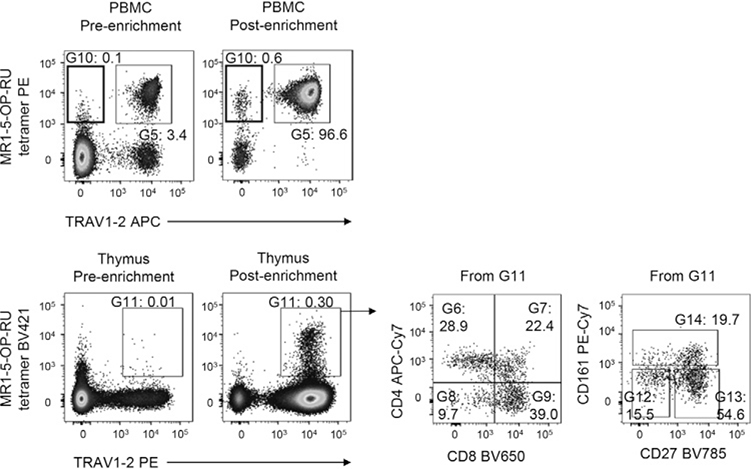
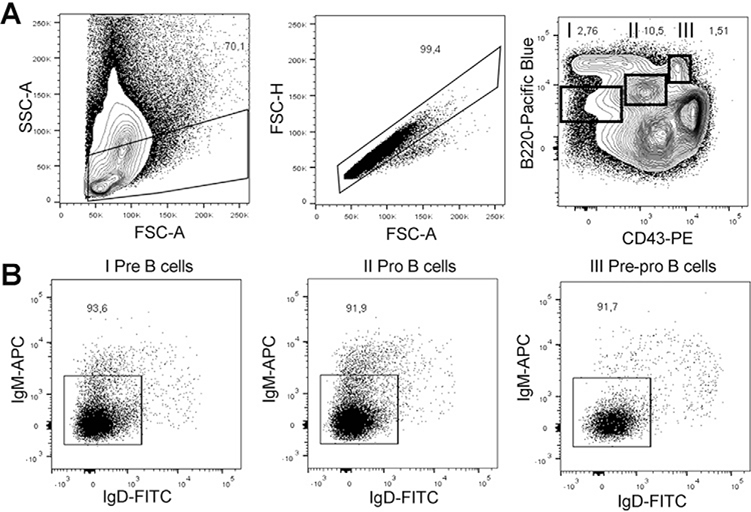
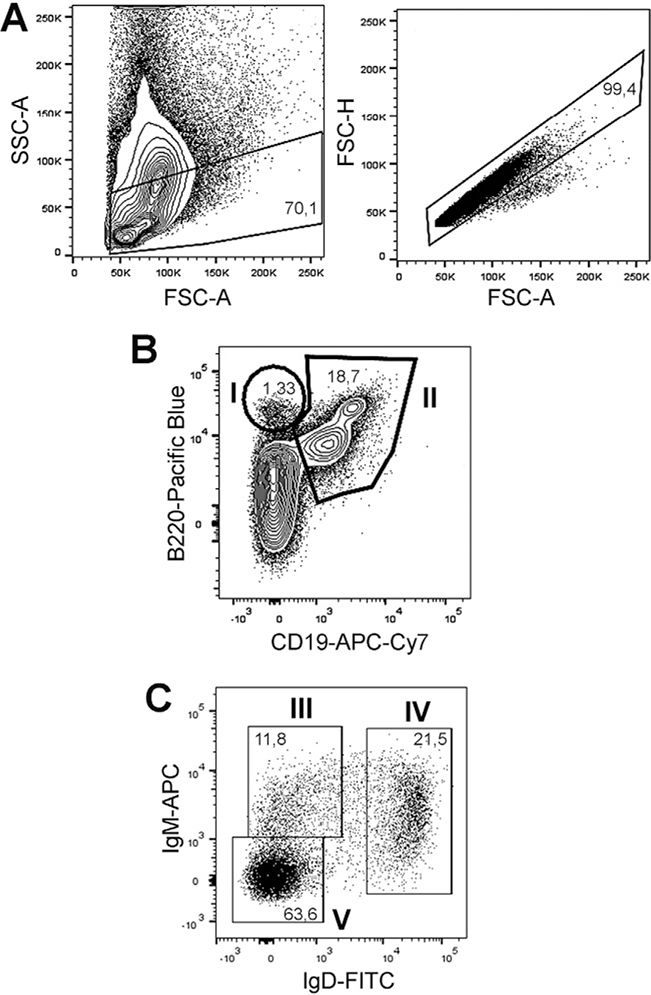
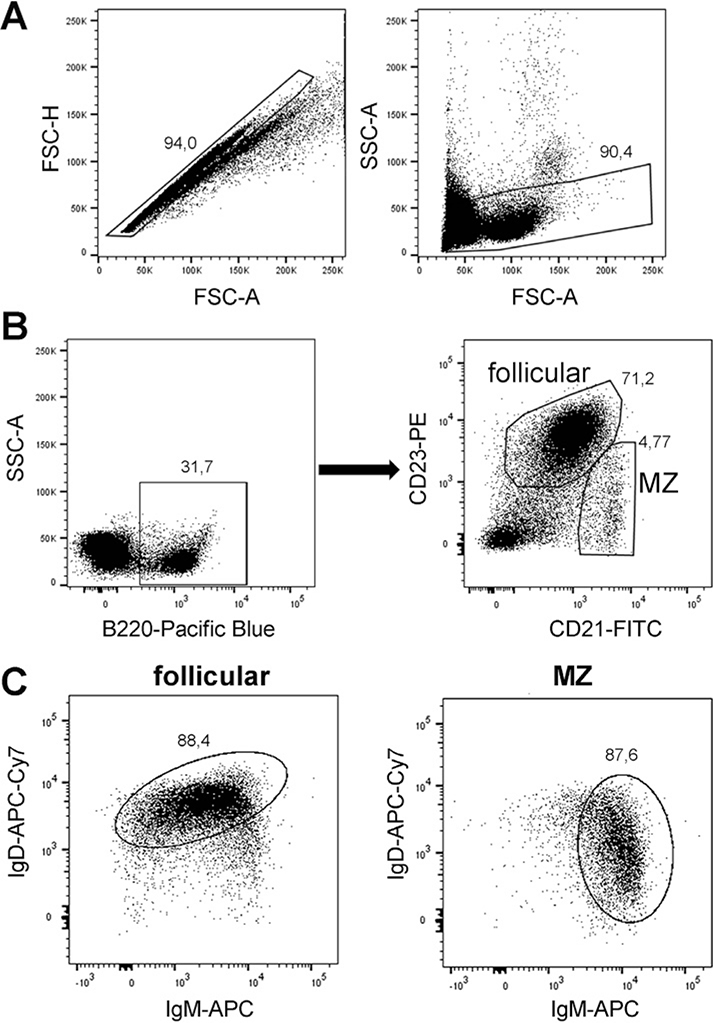
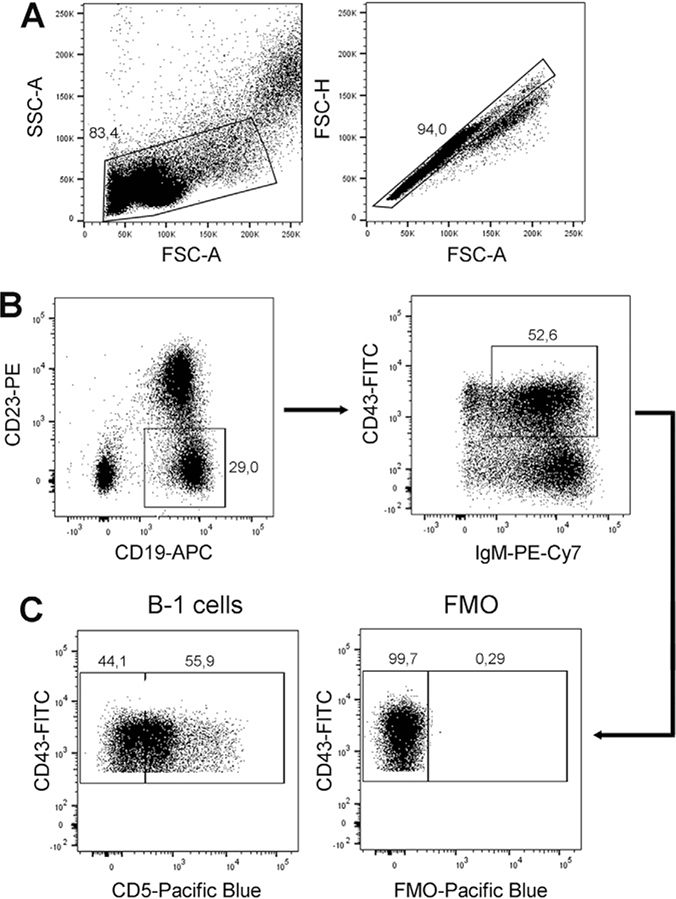
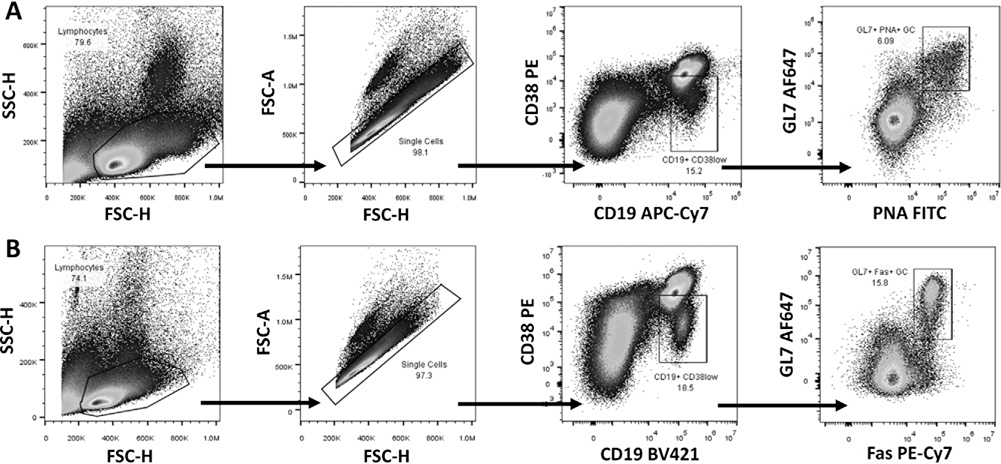
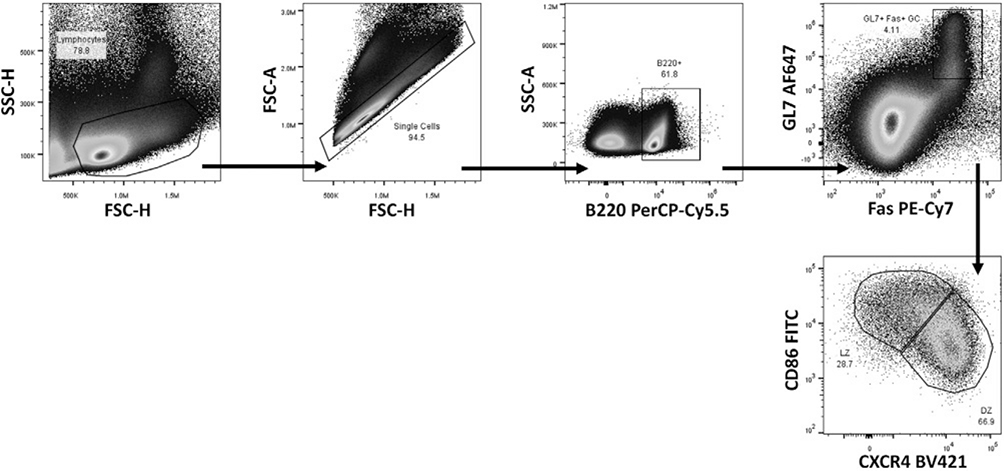
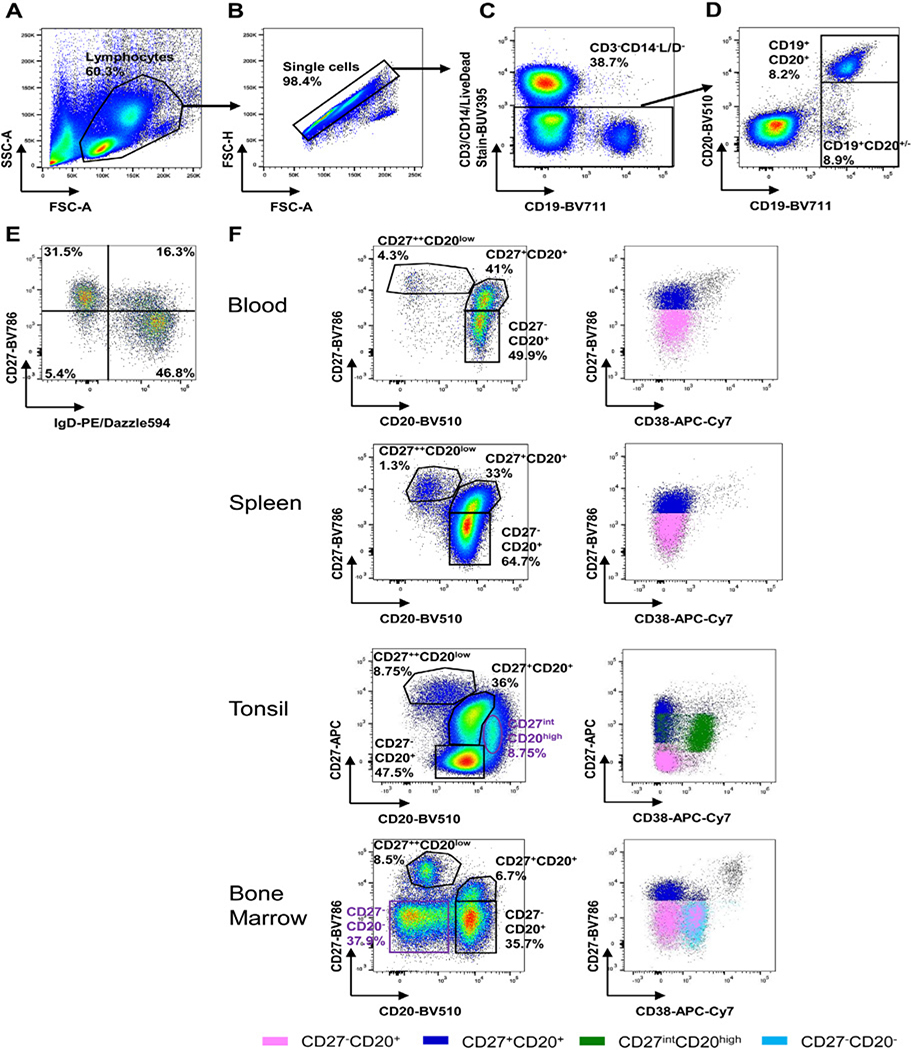
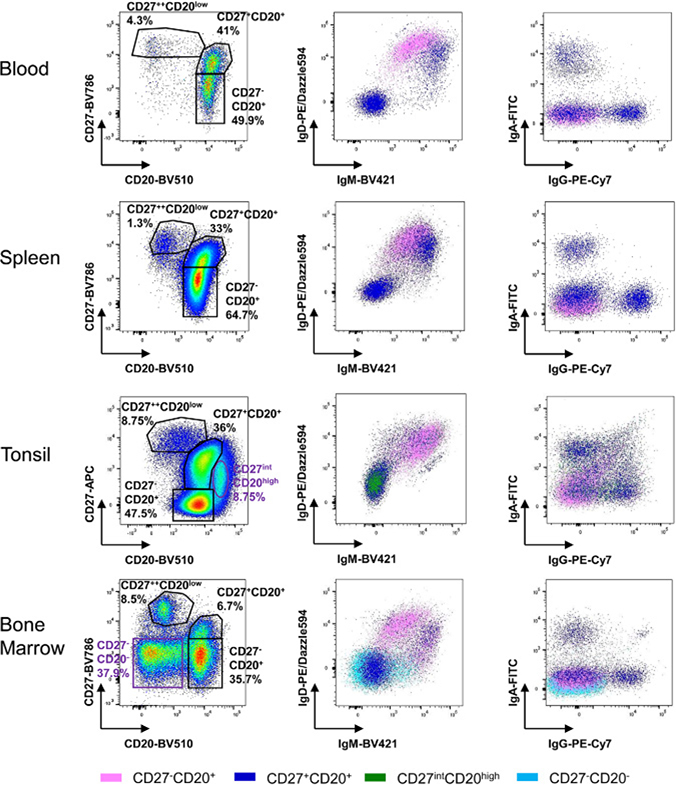
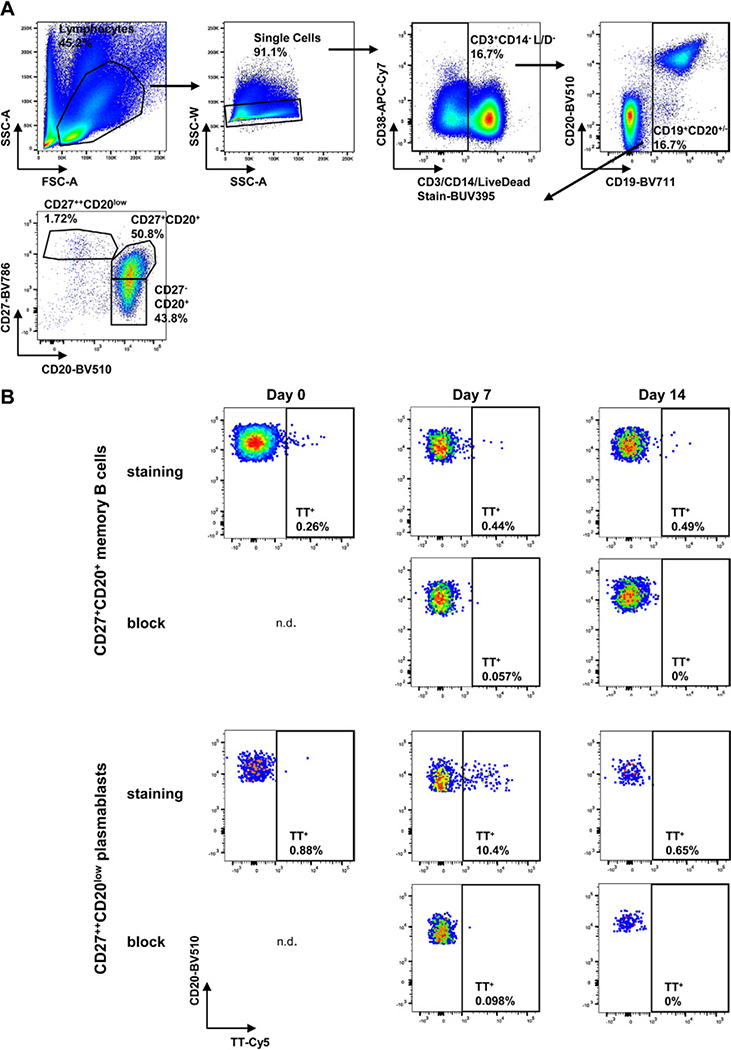
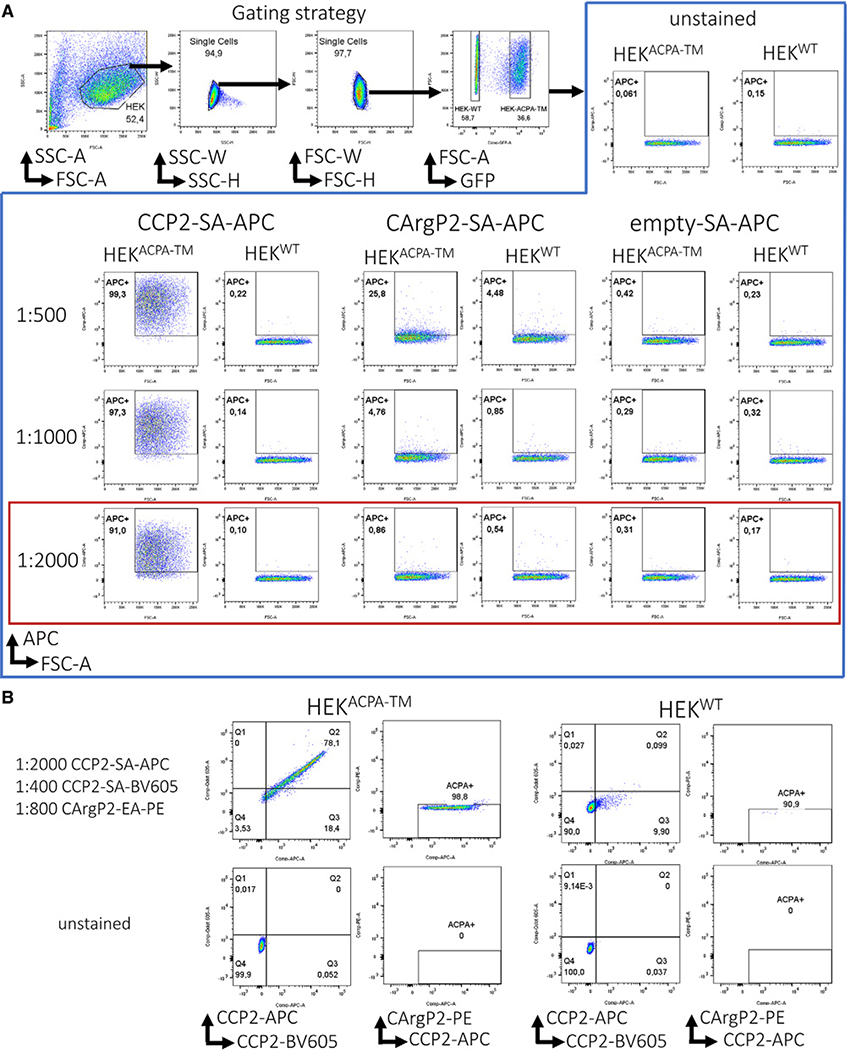
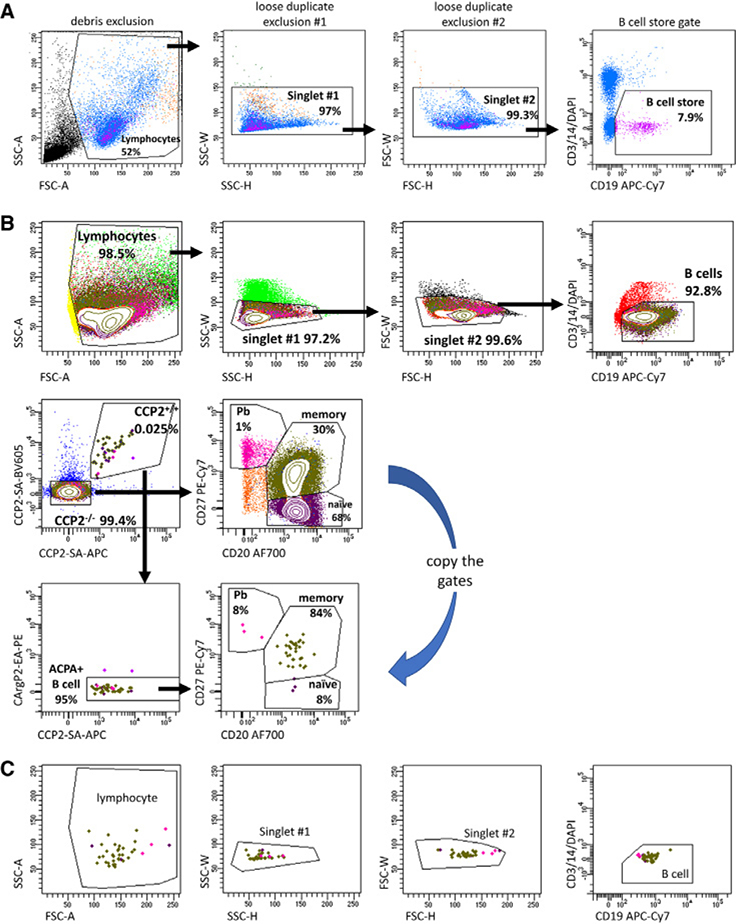
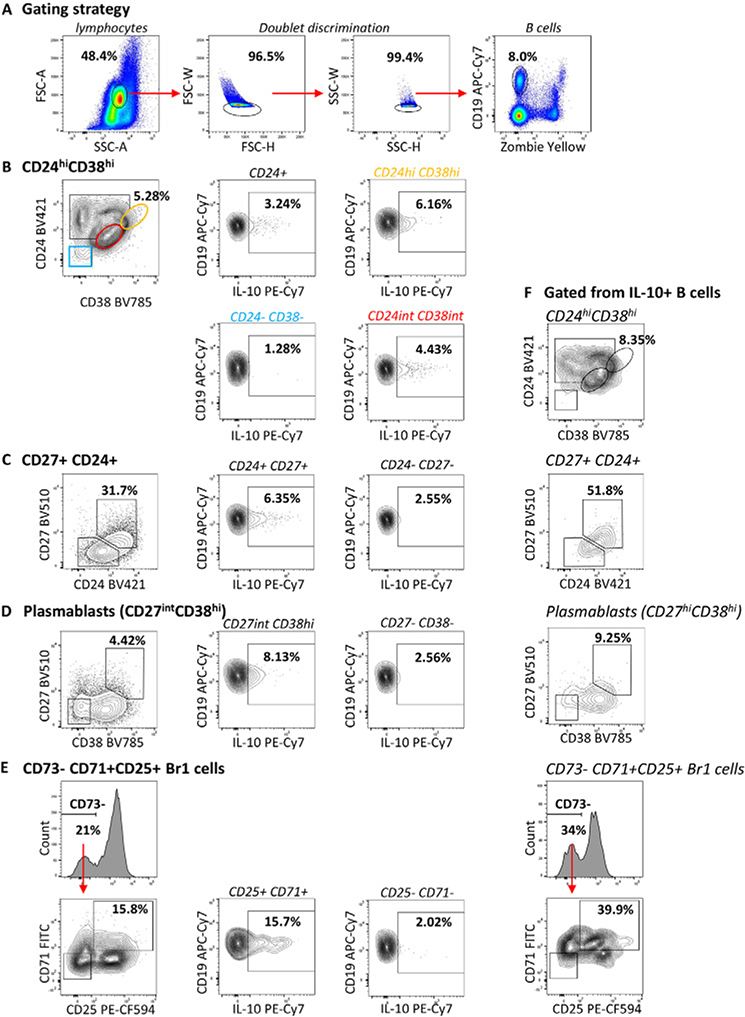
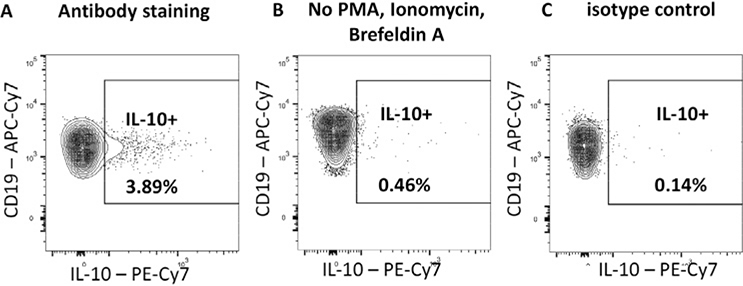
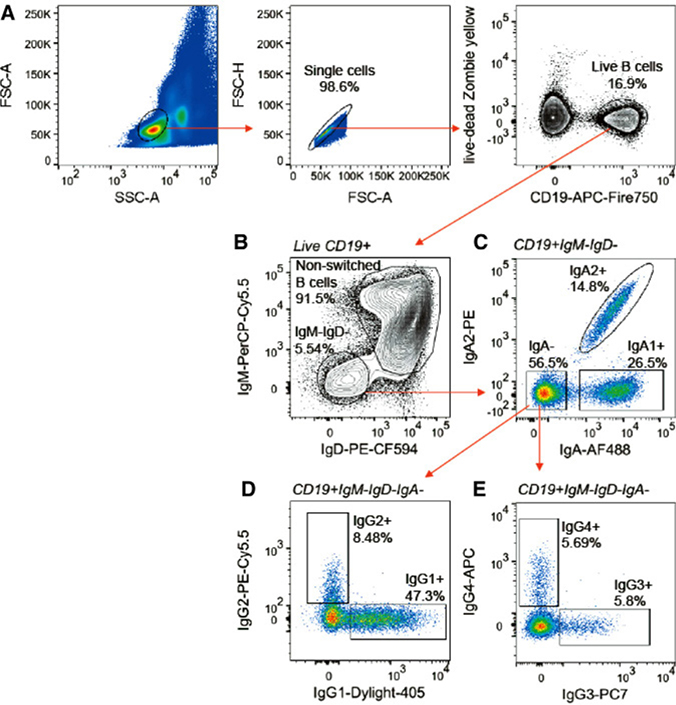

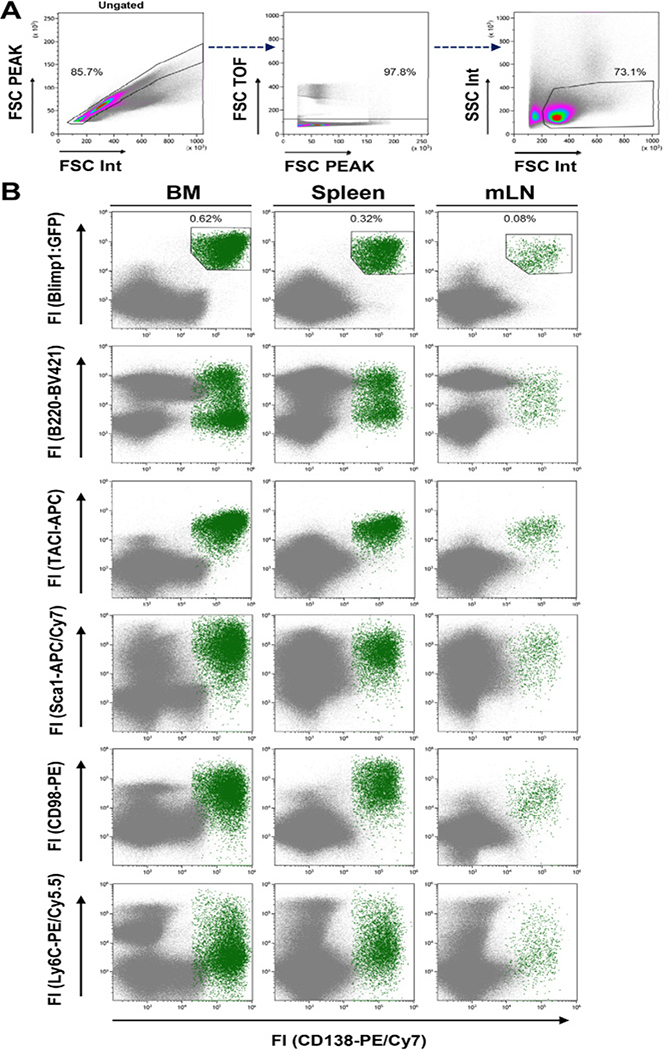

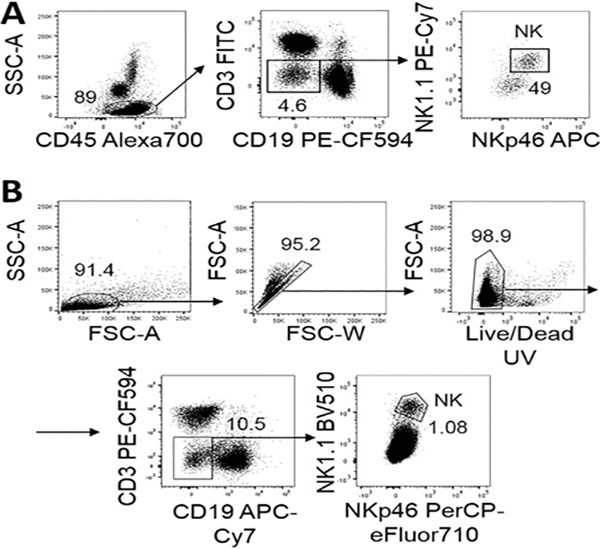
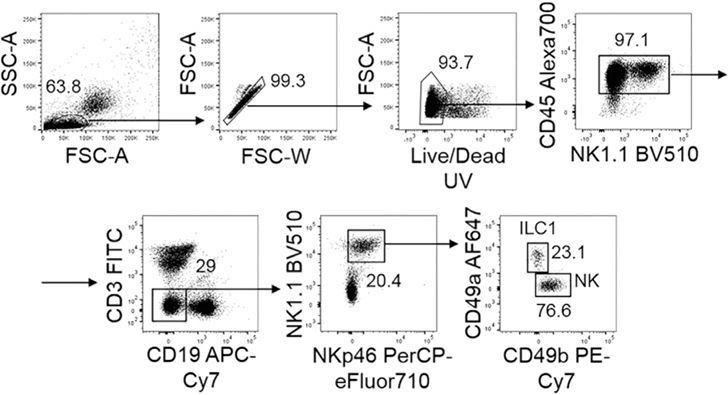

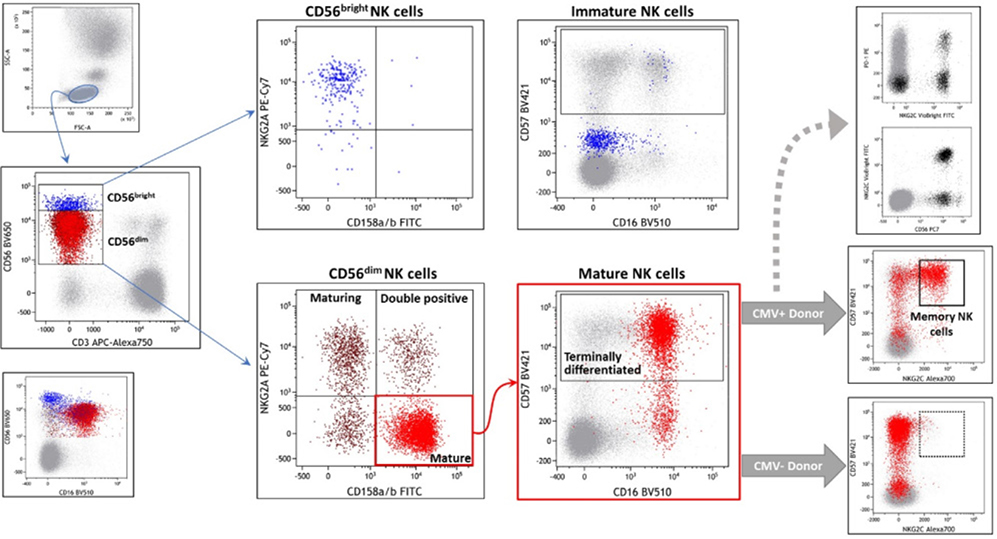
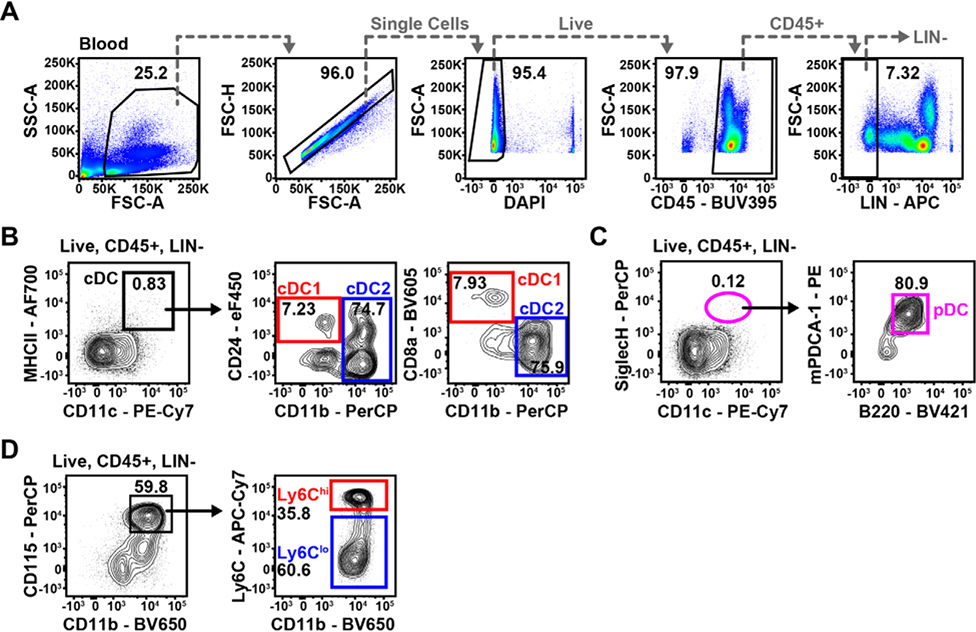
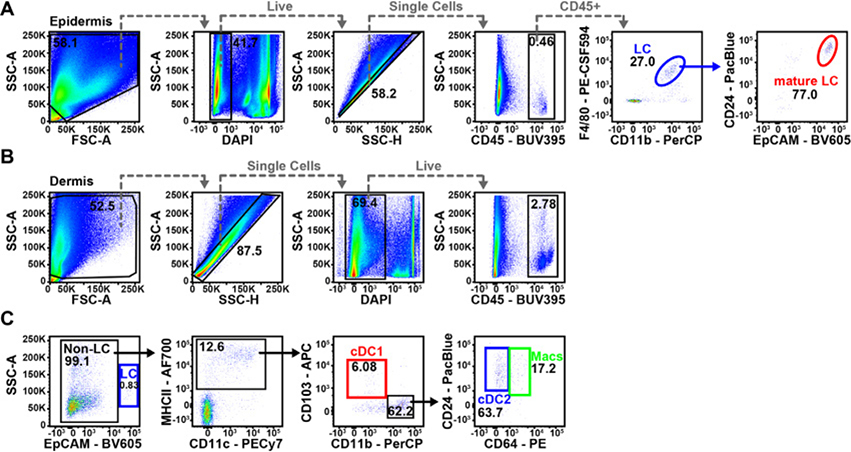
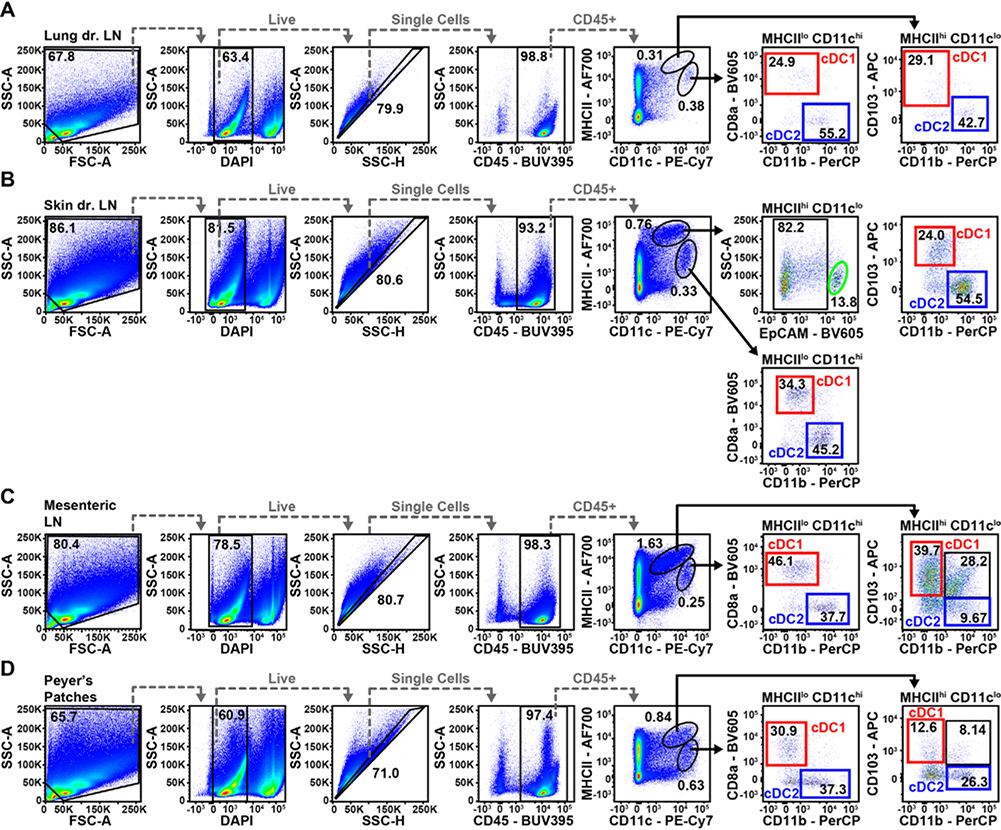
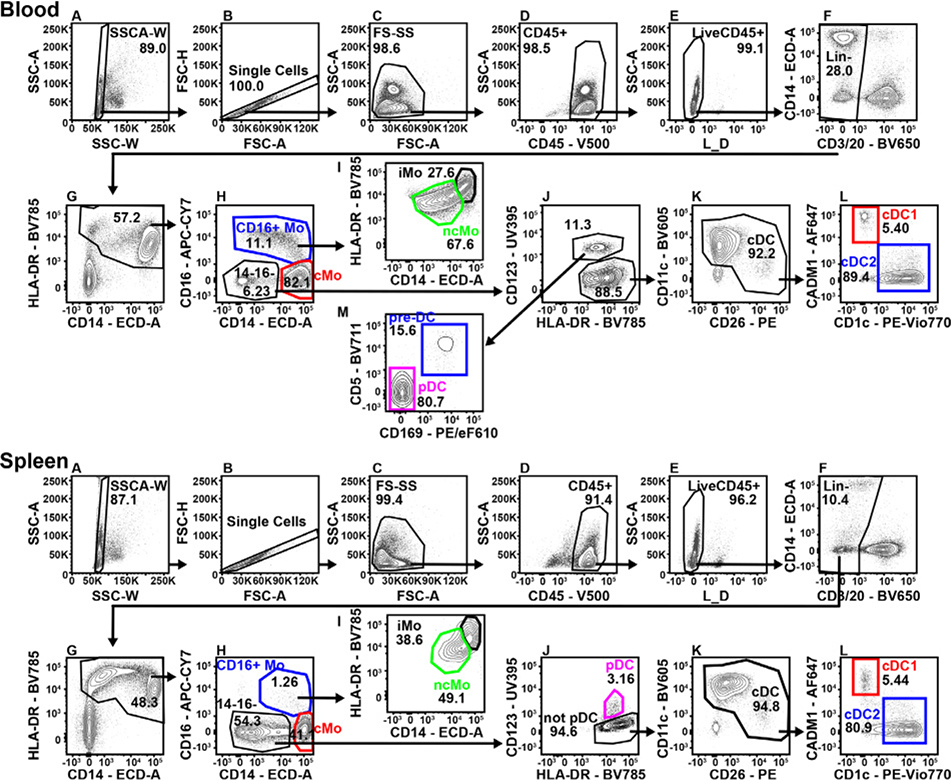

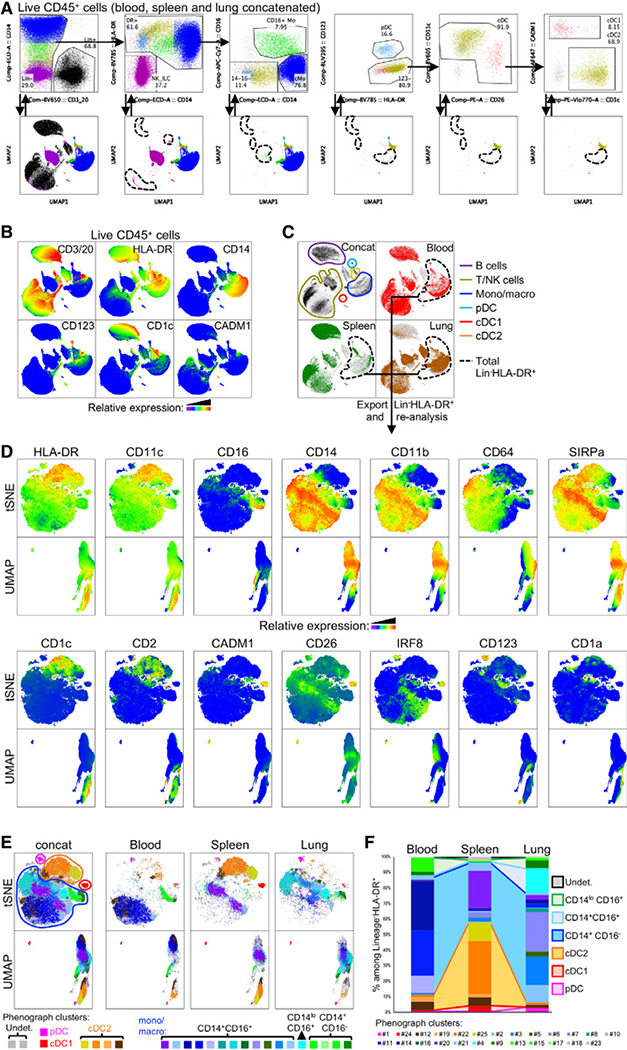
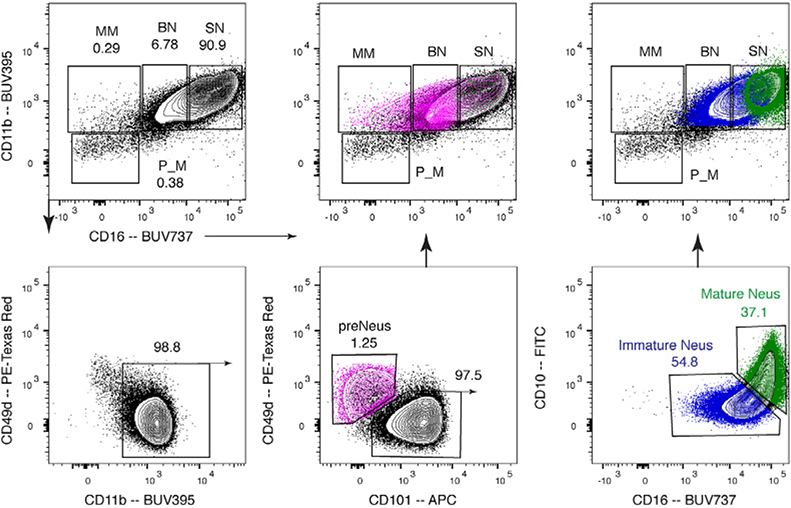
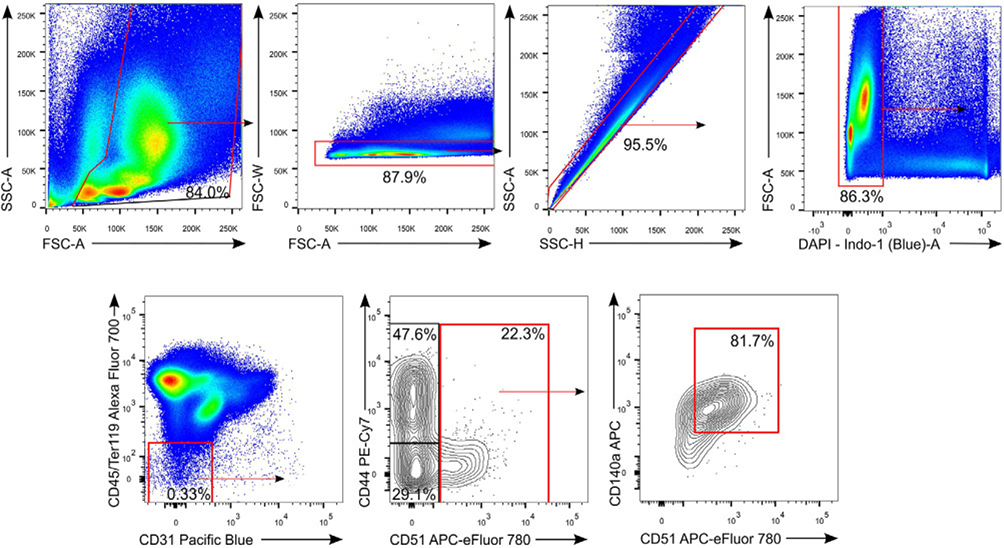
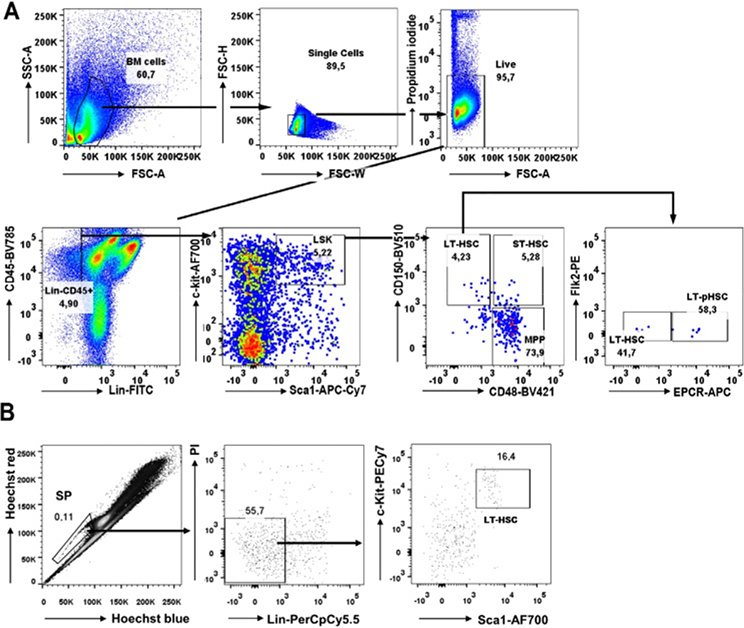
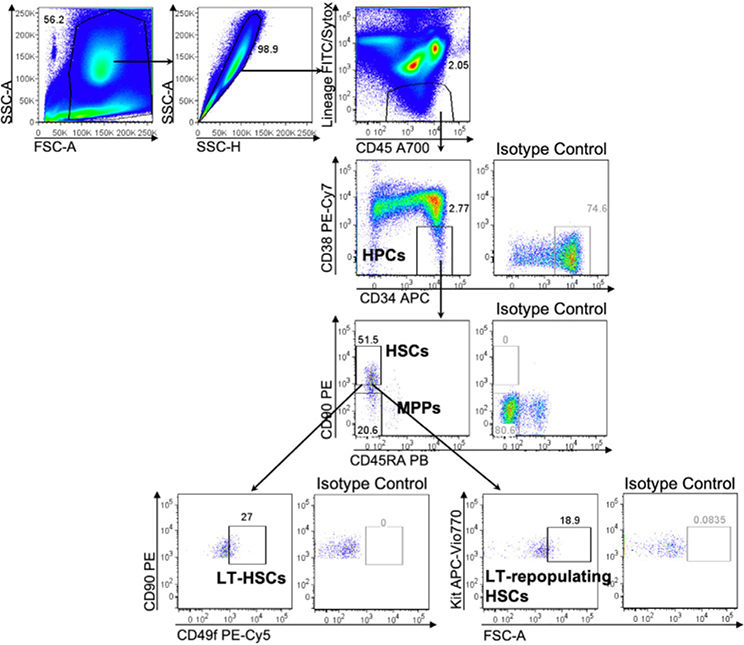
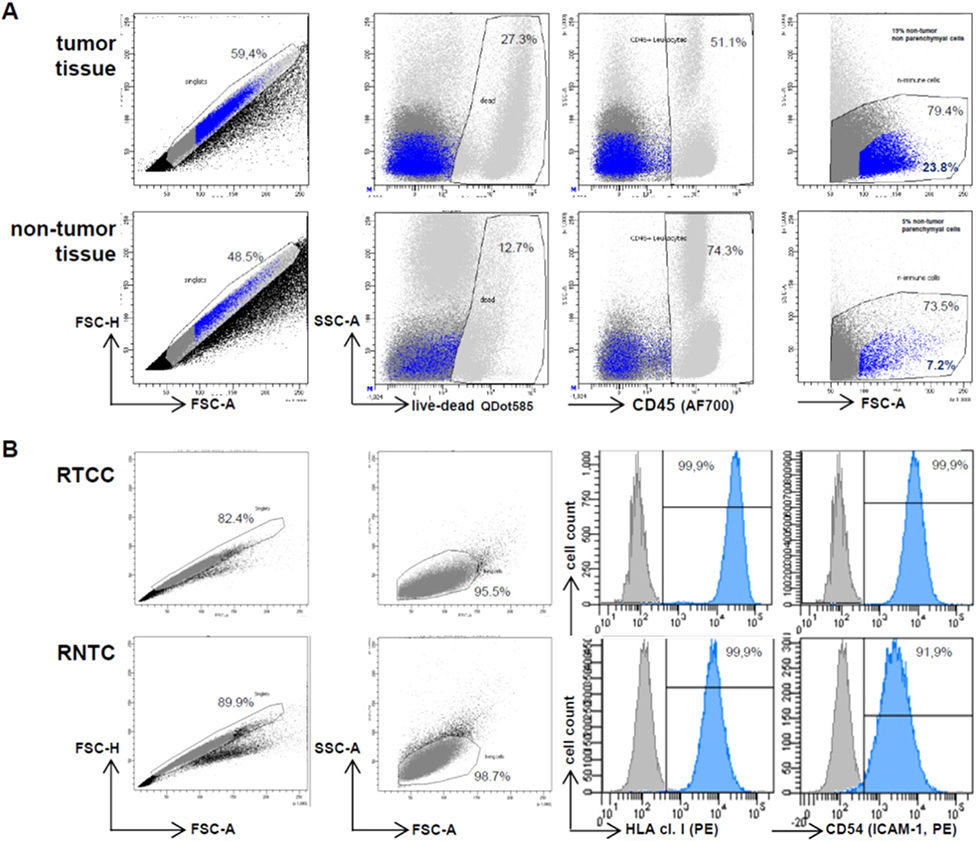
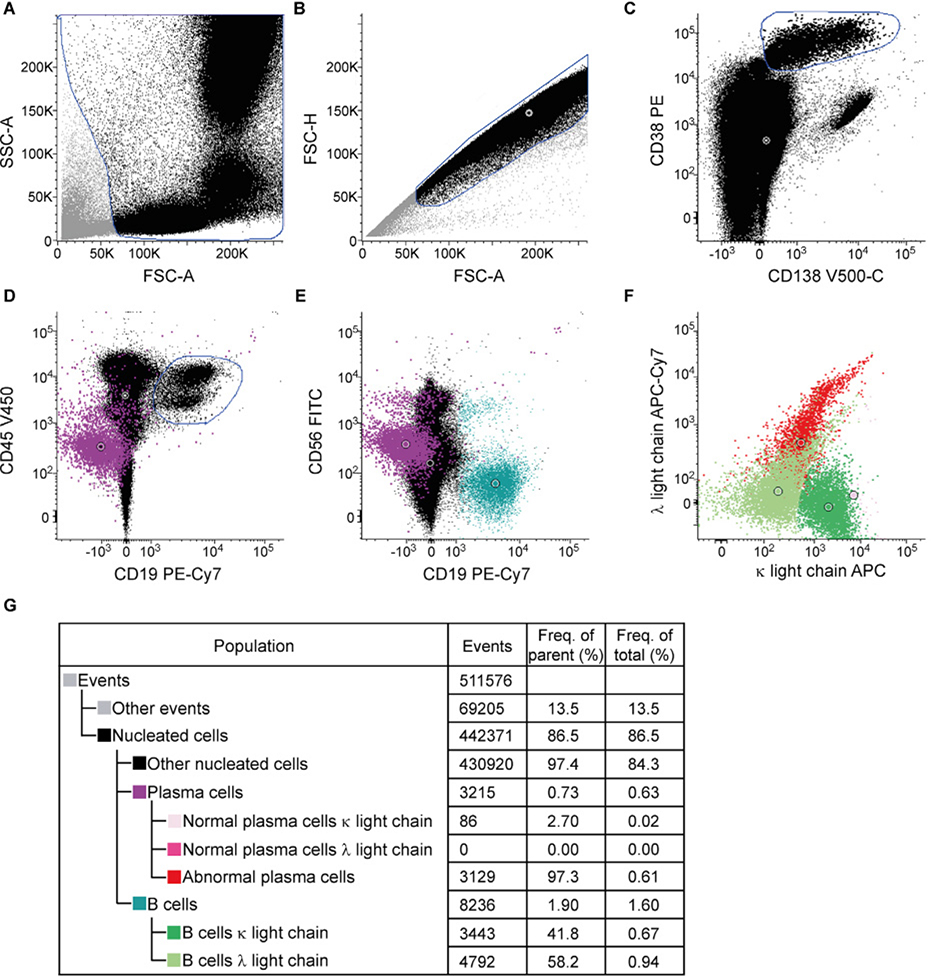
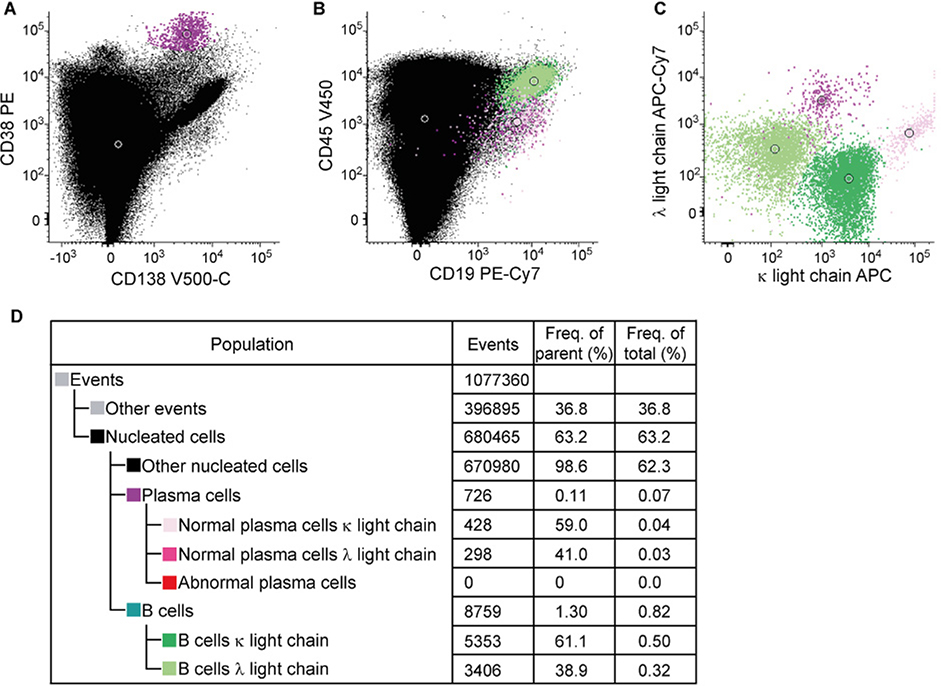
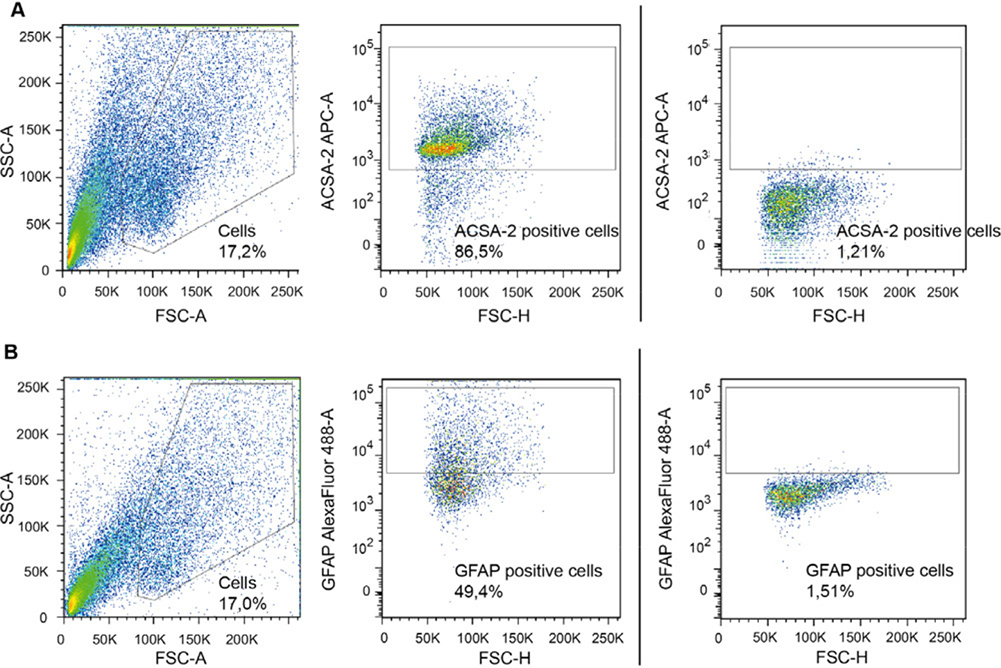
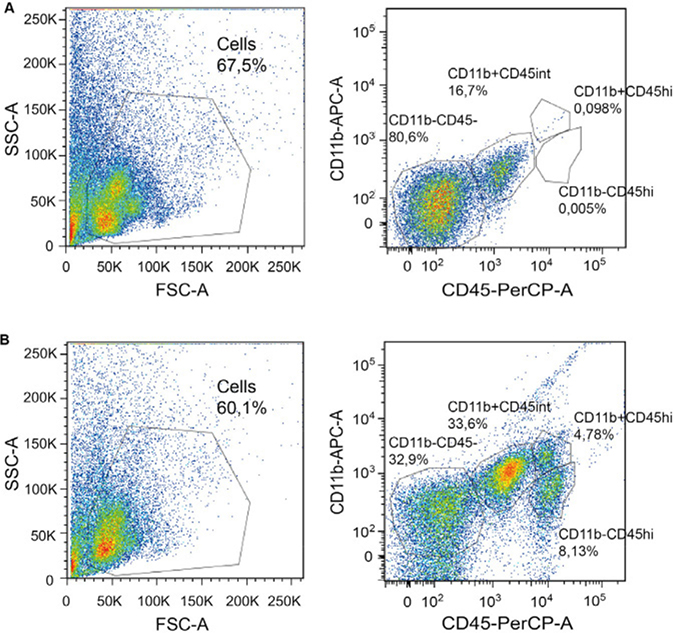
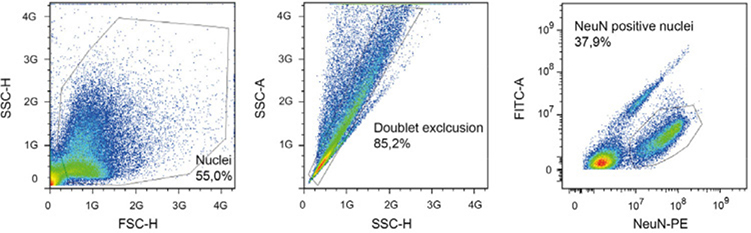
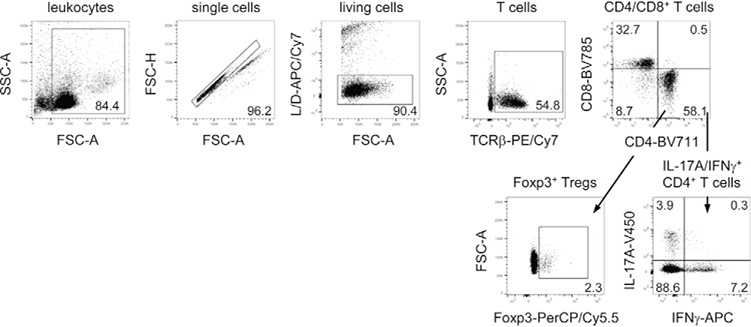
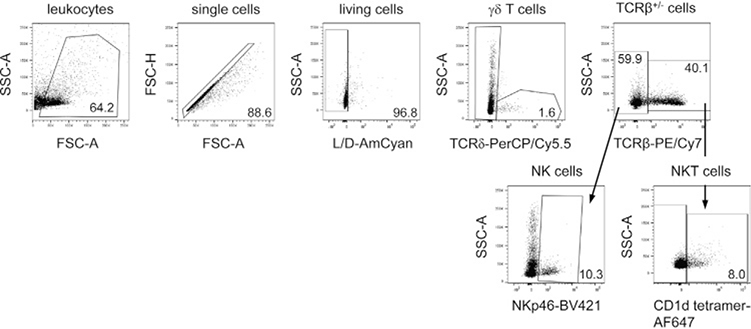
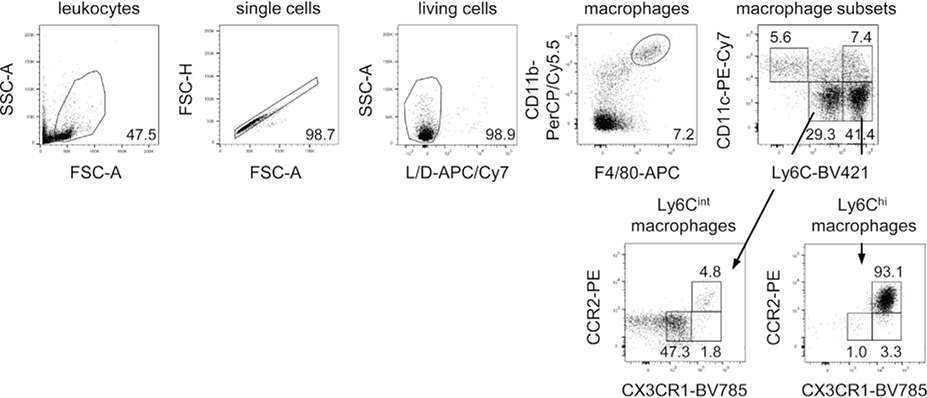

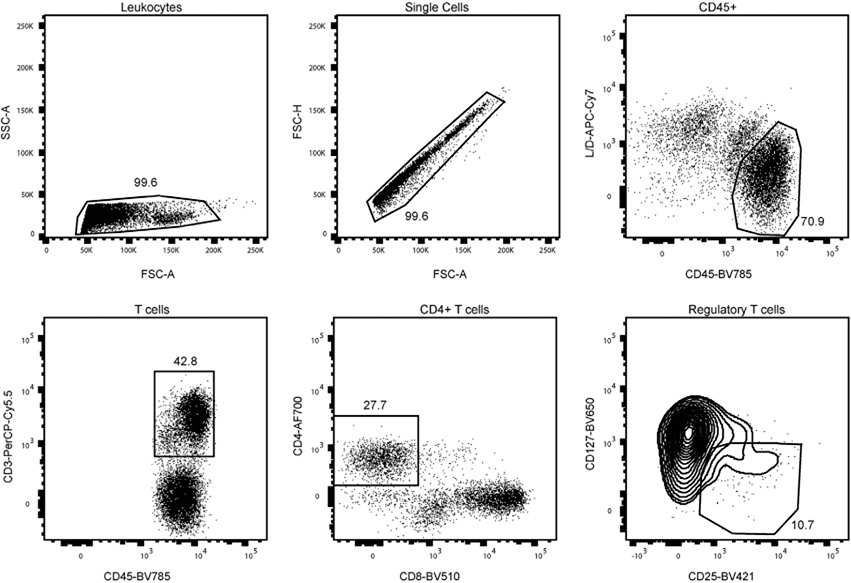
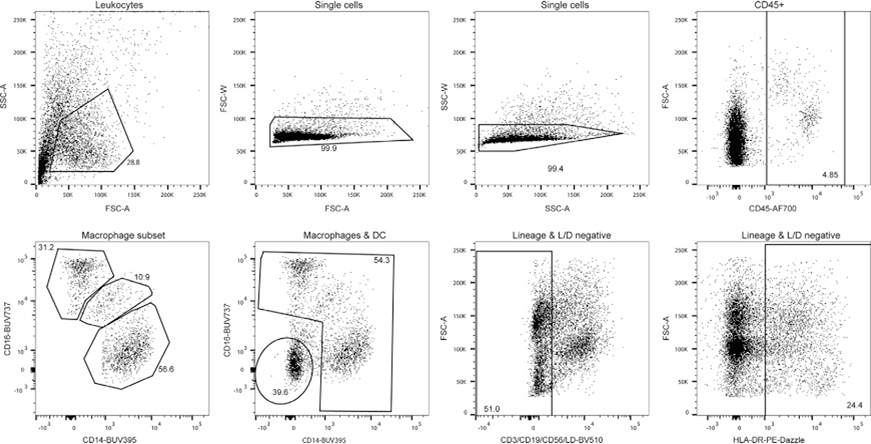
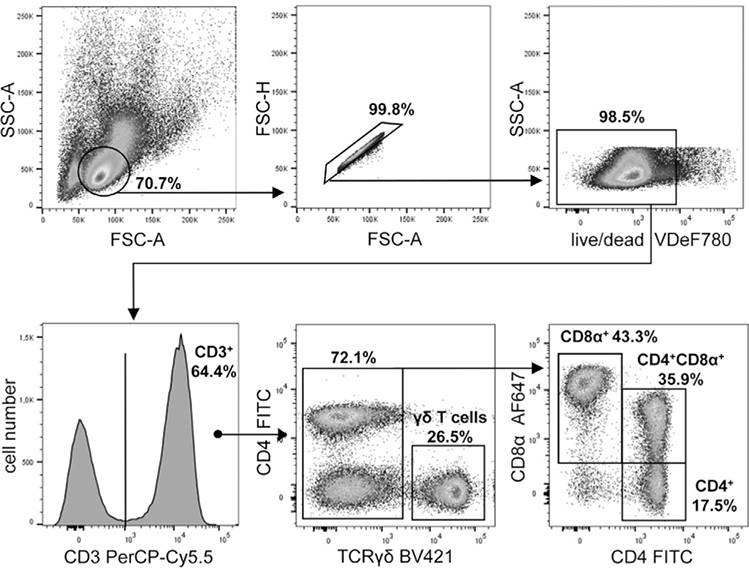
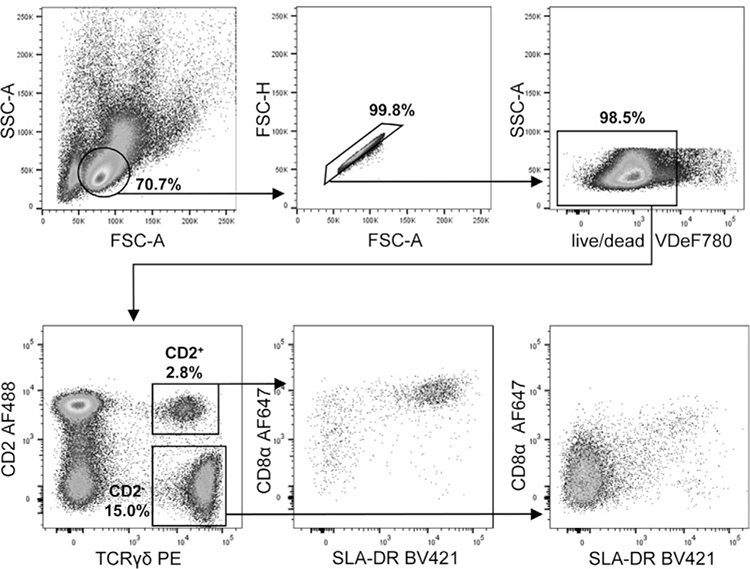
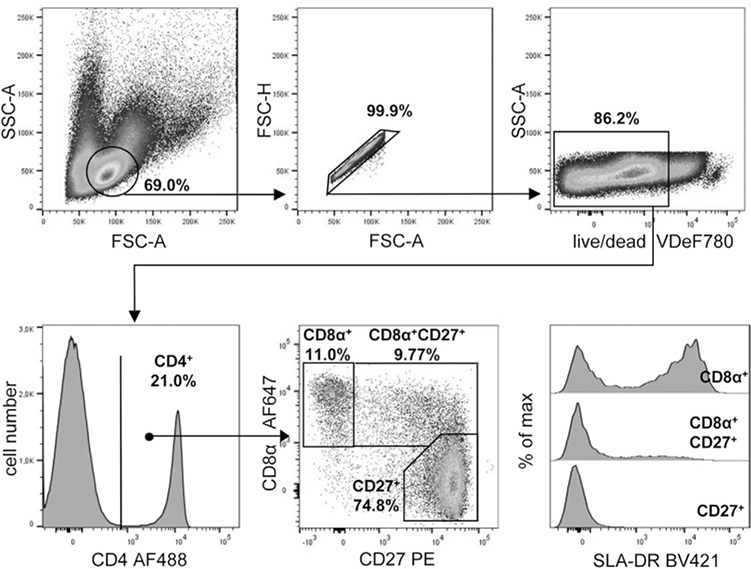
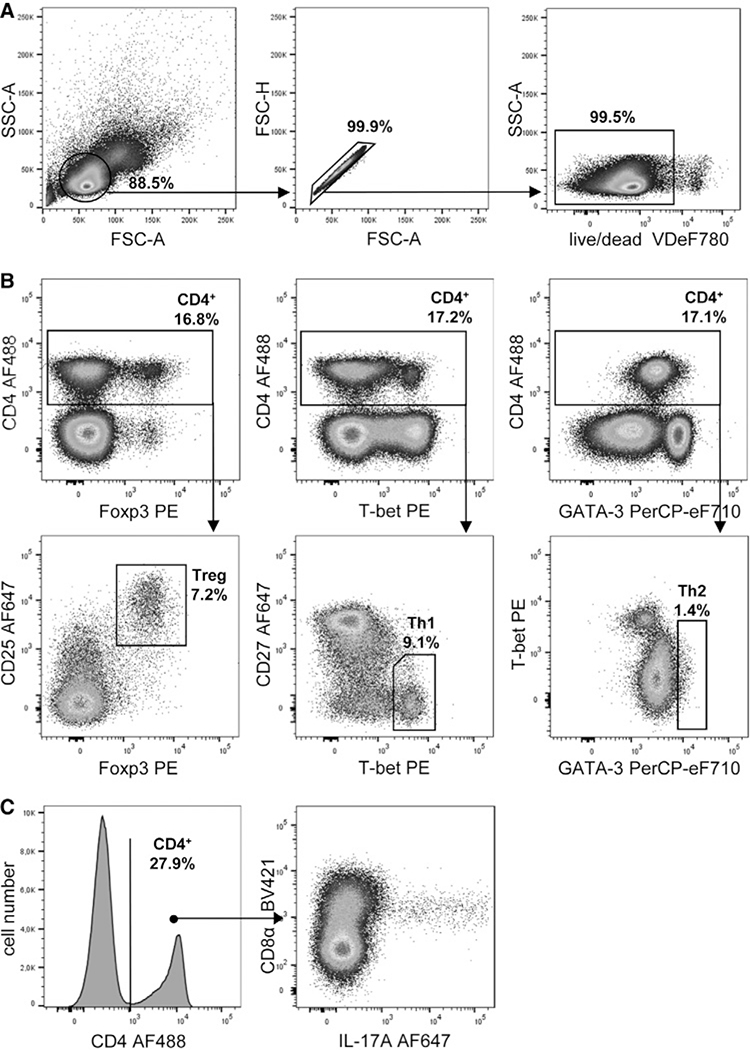
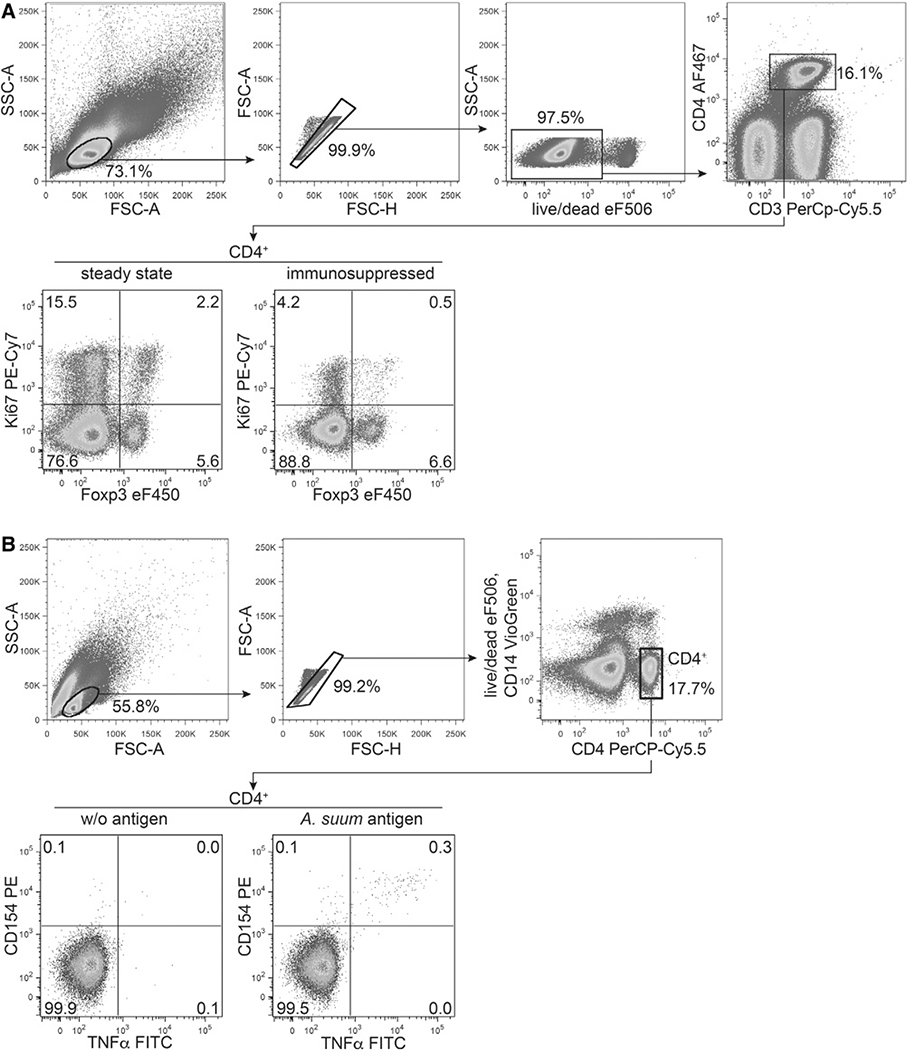


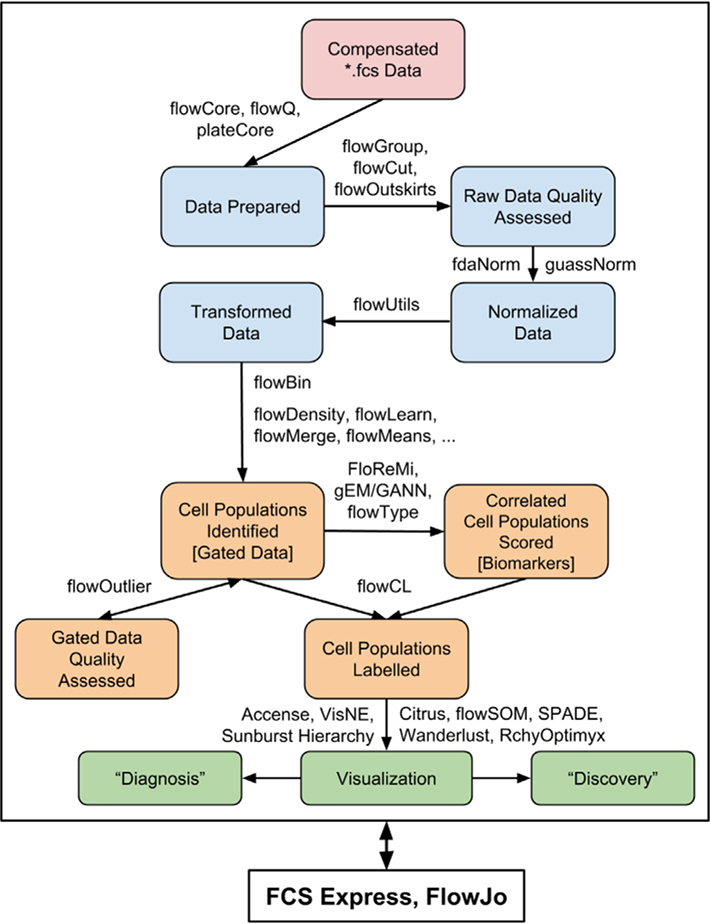
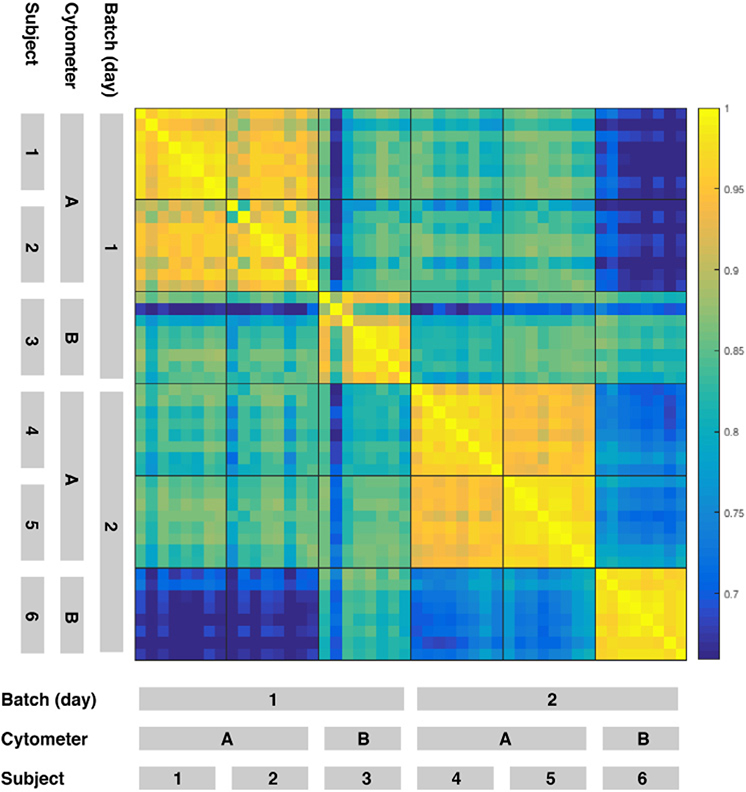
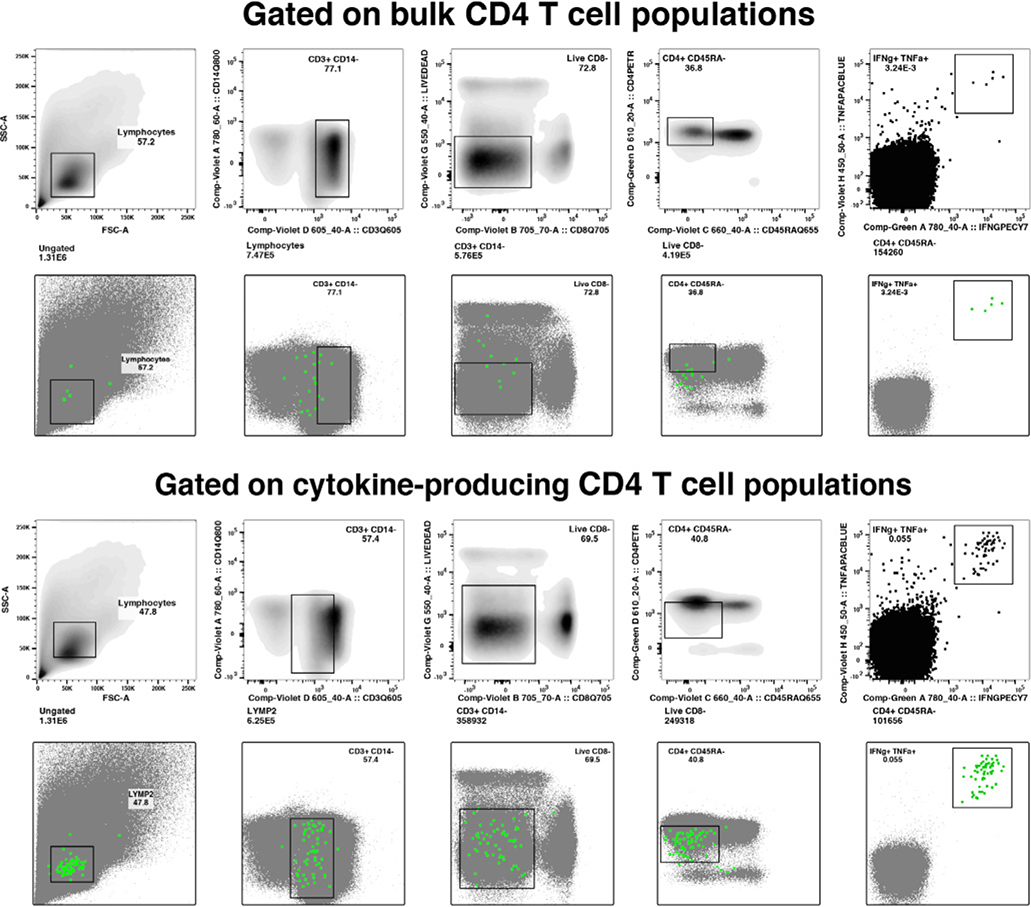
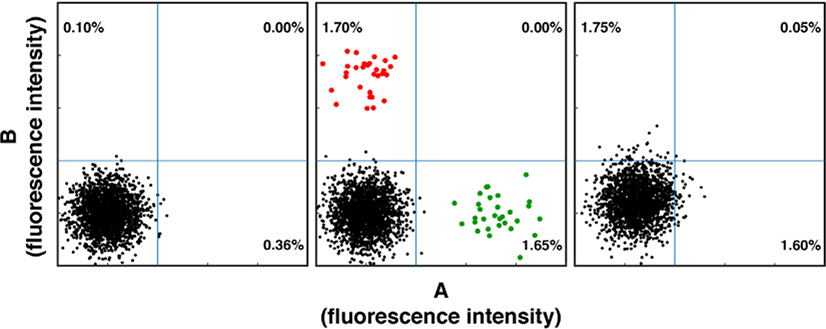



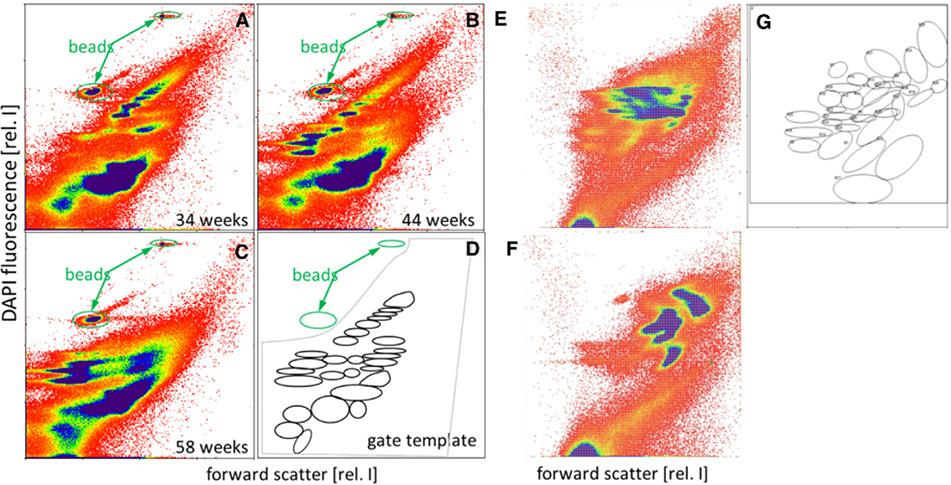
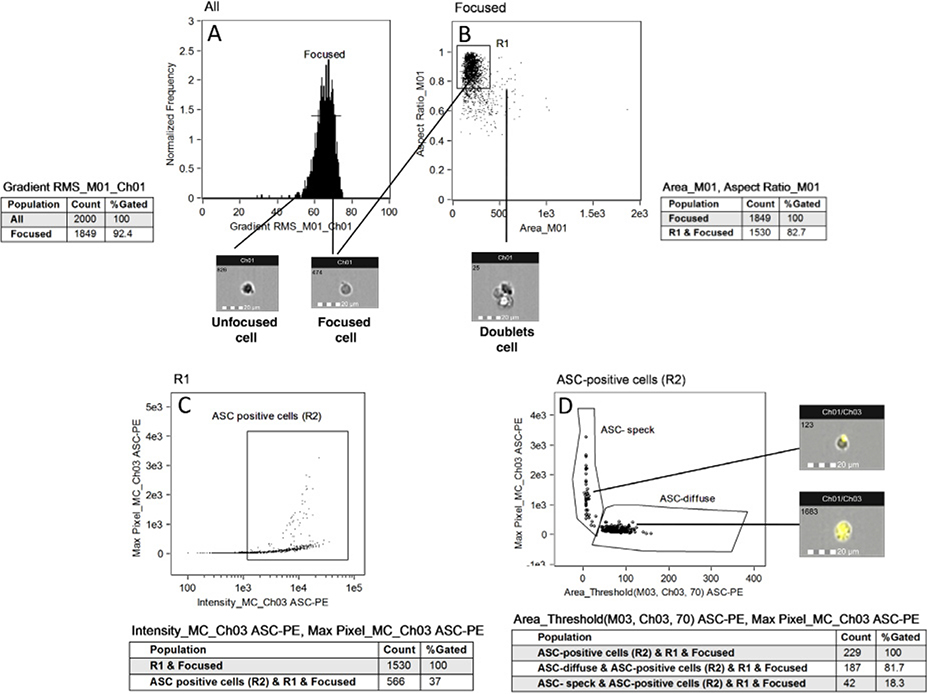
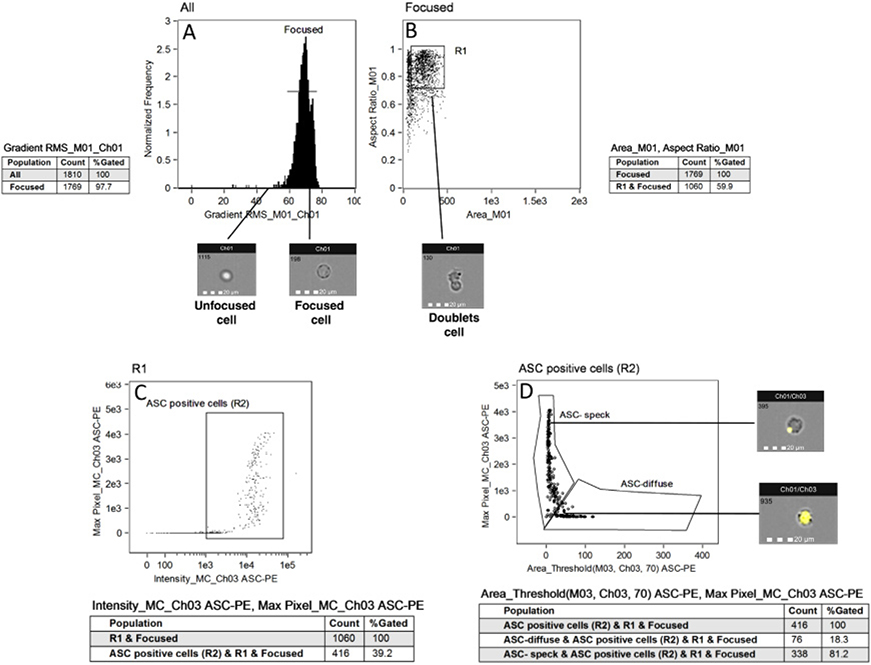
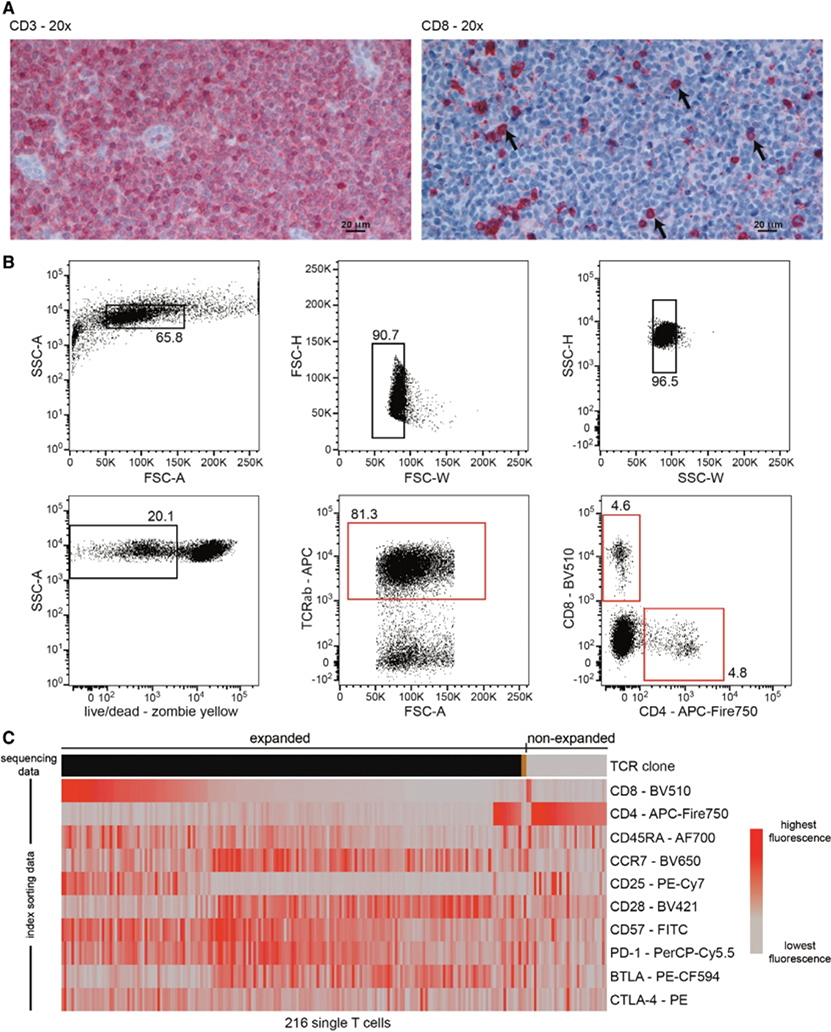
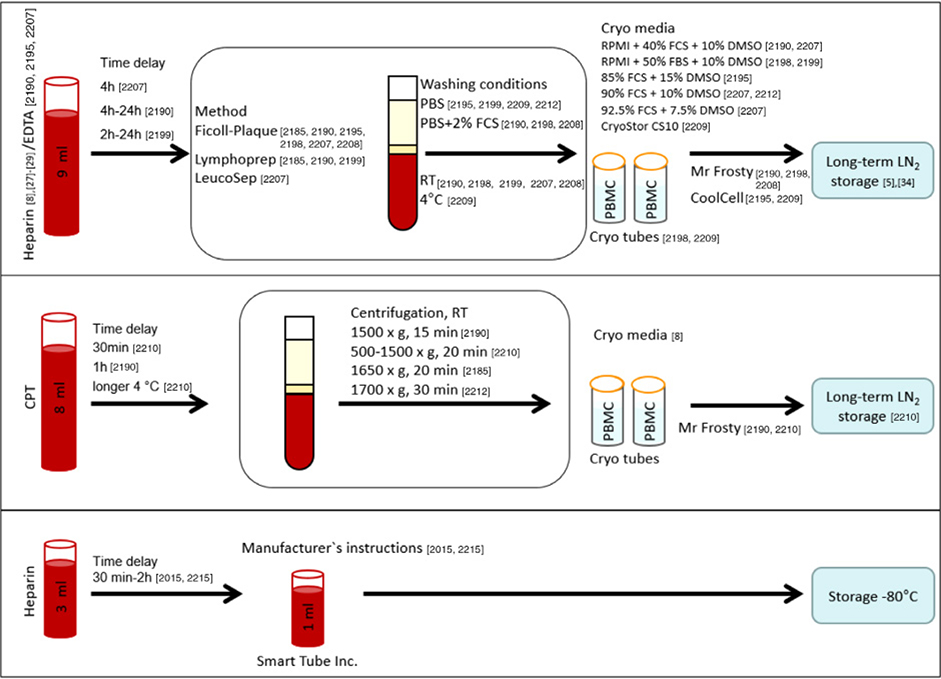
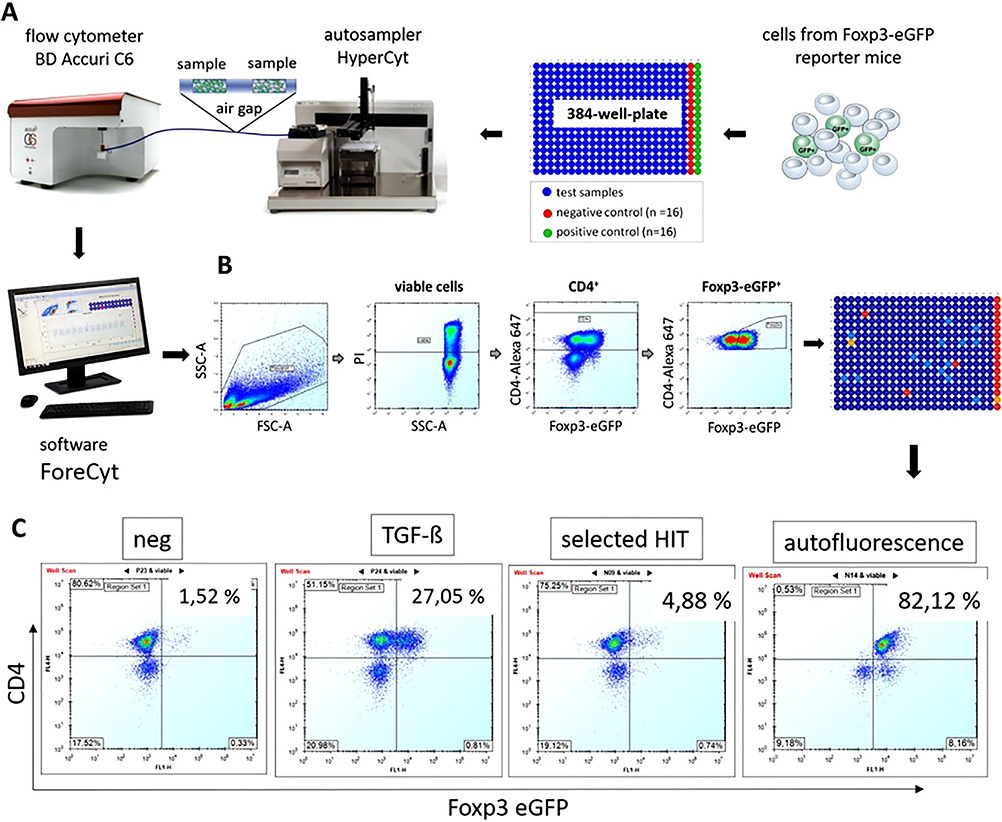
References
-
- Mack J, Fulwyler Particle Separator. US patent US 3380584 A.
-
- Kachel V, Fellner-Feldegg H and Menke E, Hydrodynamic properties of flow cytometry instruments In Melamed MR, Lindmo T and Mendelsohn ML (Eds.), Flow cytometry and sorting, 2nd ed., Wiley, New York, NY, 1990, pp. 27–44.
-
- Crosland-Taylor PJ, A device for counting small particles suspended in a fluid through a tube. Nature 1953. 171: 37–38. - PubMed
-
- Gucker FT Jr., O’Konski CT, Pickard HB and Pitts JN Jr., A photoelectronic counter for colloidal particles. J. Am. Chem. Soc 1947. 69: 2422–2431. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
- Archivio Istituzionale della Ricerca Unimi - Access Free Full Text
- Europe PubMed Central
- Institutional Repository of University of Modena and Reggio Emilia (IRIS UNIMORE)
- MLibrary (Deep Blue) - Access Free Full Text
- Ovid Technologies, Inc.
- PubMed Central
- Wiley
- eScholarship, University of California - Access Free Full Text
Other Literature Sources
Molecular Biology Databases
