

Computational Framework for High-Quality Production and Large-Scale Evolutionary Analysis of Metagenome Assembled Genomes
- PMID: 31633780
- PMCID: PMC6993843
- DOI: 10.1093/molbev/msz237
Computational Framework for High-Quality Production and Large-Scale Evolutionary Analysis of Metagenome Assembled Genomes
Abstract
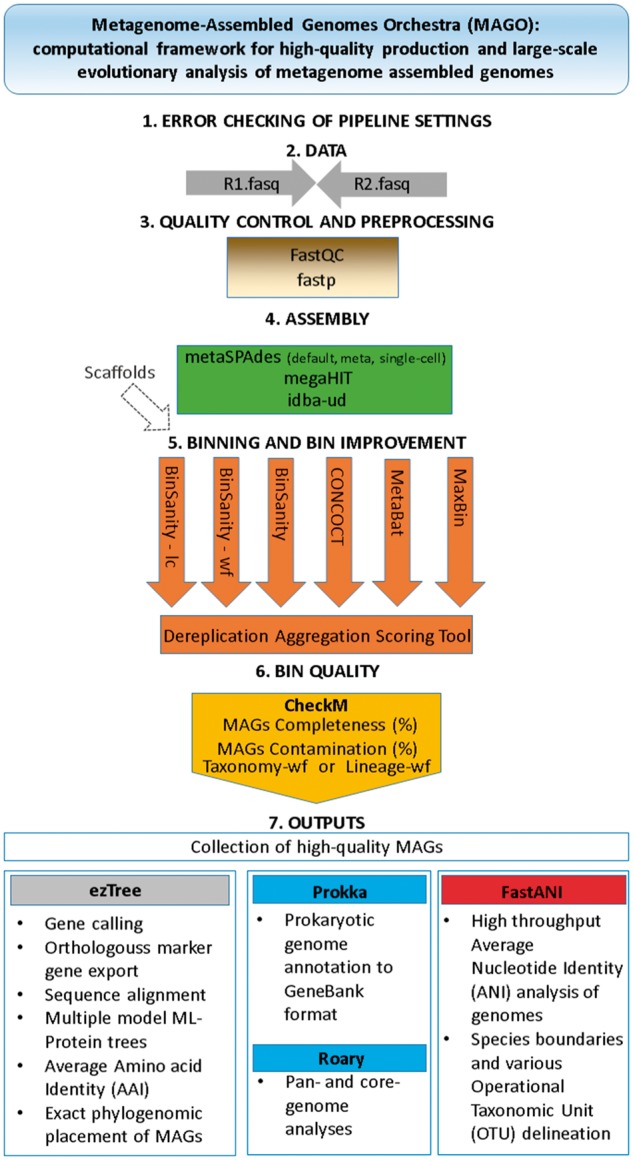
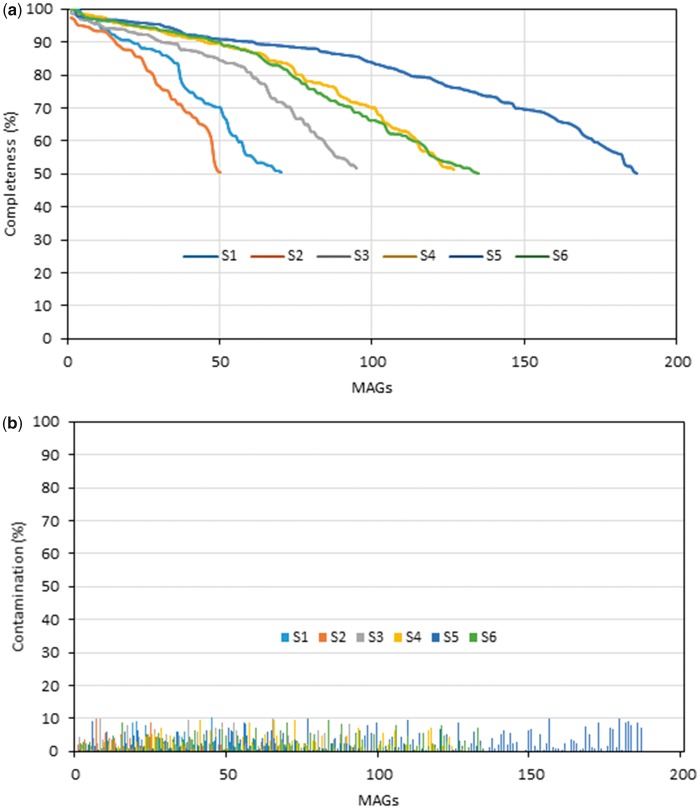
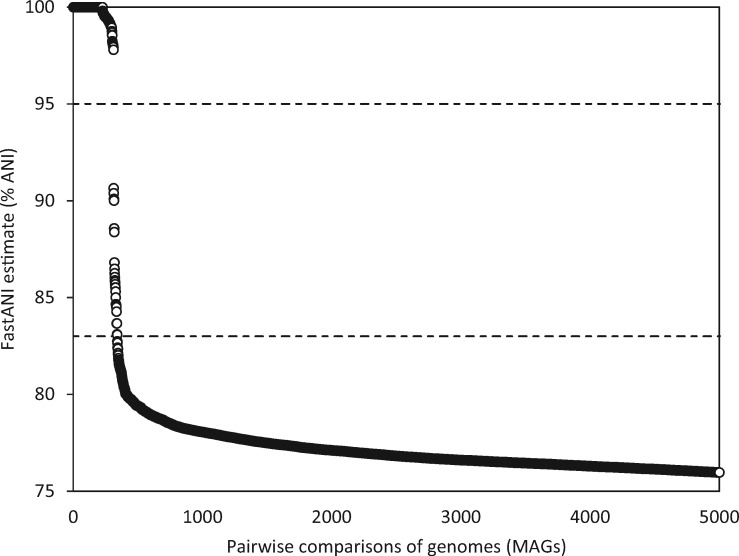
Microbial species play important roles in different environments and the production of high-quality genomes from metagenome data sets represents a major obstacle to understanding their ecological and evolutionary dynamics. Metagenome-Assembled Genomes Orchestra (MAGO) is a computational framework that integrates and simplifies metagenome assembly, binning, bin improvement, bin quality (completeness and contamination), bin annotation, and evolutionary placement of bins via detailed maximum-likelihood phylogeny based on multiple marker genes using different amino acid substitution models, next to average nucleotide identity analysis of genomes for delineation of species boundaries and operational taxonomic units. MAGO offers streamlined execution of the entire metagenomics pipeline, error checking, computational resource distribution and compatibility of data formats, governed by user-tailored pipeline processing. MAGO is an open-source-software package released in three different ways, as a singularity image and a Docker container for HPC purposes as well as for running MAGO on a commodity hardware, and a virtual machine for gaining a full access to MAGO underlying structure and source code. MAGO is open to suggestions for extensions and is amenable for use in both research and teaching of genomics and molecular evolution of genomes assembled from small single-cell projects or large-scale and complex environmental metagenomes.
Keywords: FastANI; evolutionary analyses; genome assembly and binning; metagenomics; microbial draft genomes; species boundaries.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C.. 2014. Binning metagenomics contigs by coverage and composition. Nat Methods. 11(11):1144–1146. - PubMed
-
- Andrews A. 2010. FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; last accessed September 04, 2019.
-
- Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 35(8):725–731. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
