A comprehensive study of metabolite genetics reveals strong pleiotropy and heterogeneity across time and context
- PMID: 31636271
- PMCID: PMC6803661
- DOI: 10.1038/s41467-019-12703-7
A comprehensive study of metabolite genetics reveals strong pleiotropy and heterogeneity across time and context
Abstract
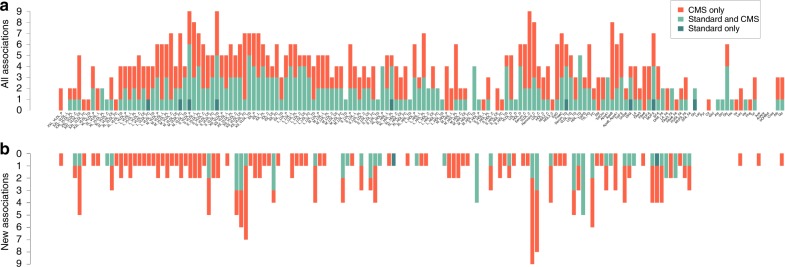
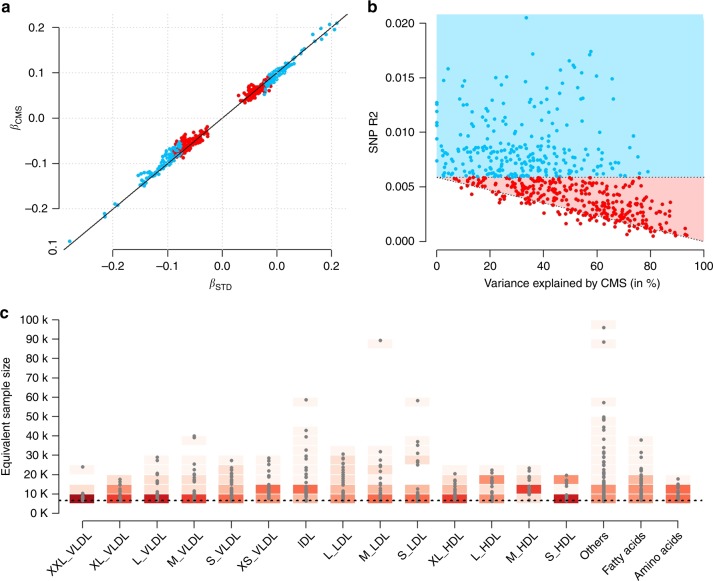
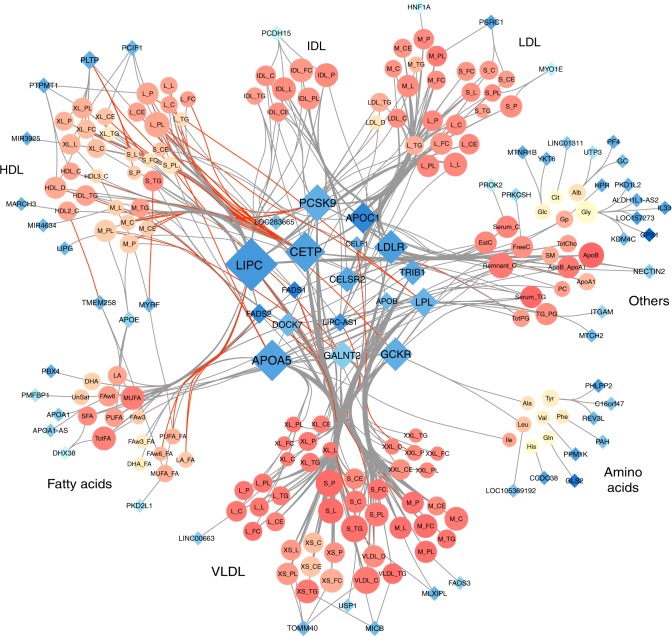
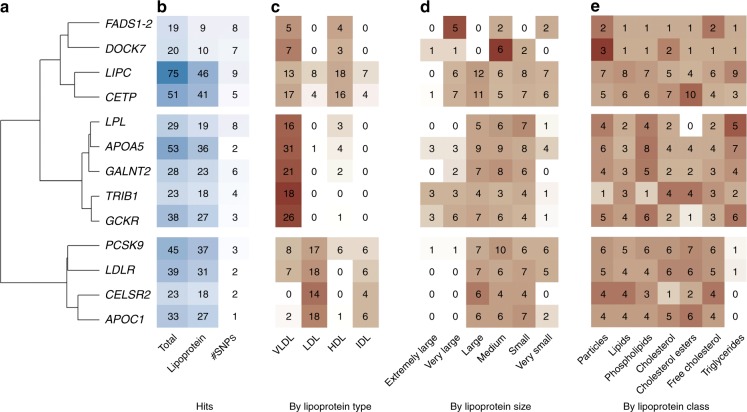
Genetic studies of metabolites have identified thousands of variants, many of which are associated with downstream metabolic and obesogenic disorders. However, these studies have relied on univariate analyses, reducing power and limiting context-specific understanding. Here we aim to provide an integrated perspective of the genetic basis of metabolites by leveraging the Finnish Metabolic Syndrome In Men (METSIM) cohort, a unique genetic resource which contains metabolic measurements, mostly lipids, across distinct time points as well as information on statin usage. We increase effective sample size by an average of two-fold by applying the Covariates for Multi-phenotype Studies (CMS) approach, identifying 588 significant SNP-metabolite associations, including 228 new associations. Our analysis pinpoints a small number of master metabolic regulator genes, balancing the relative proportion of dozens of metabolite levels. We further identify associations to changes in metabolic levels across time as well as genetic interactions with statin at both the master metabolic regulator and genome-wide level.
Conflict of interest statement
The authors declare no competing interests.
Figures