

Mitochondrial dysfunction increases pro-inflammatory cytokine production and impairs repair and corticosteroid responsiveness in lung epithelium
- PMID: 31636329
- PMCID: PMC6803636
- DOI: 10.1038/s41598-019-51517-x
Mitochondrial dysfunction increases pro-inflammatory cytokine production and impairs repair and corticosteroid responsiveness in lung epithelium
Abstract
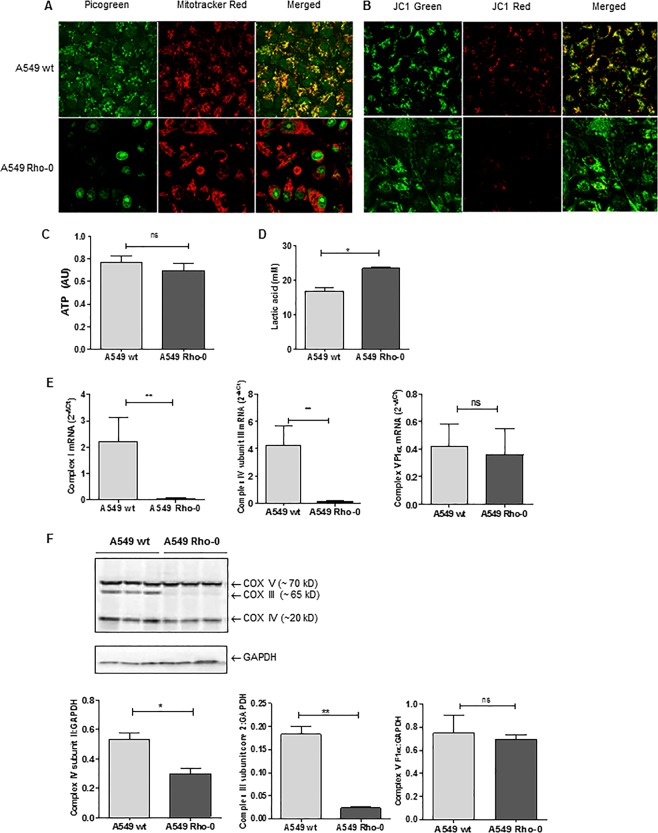
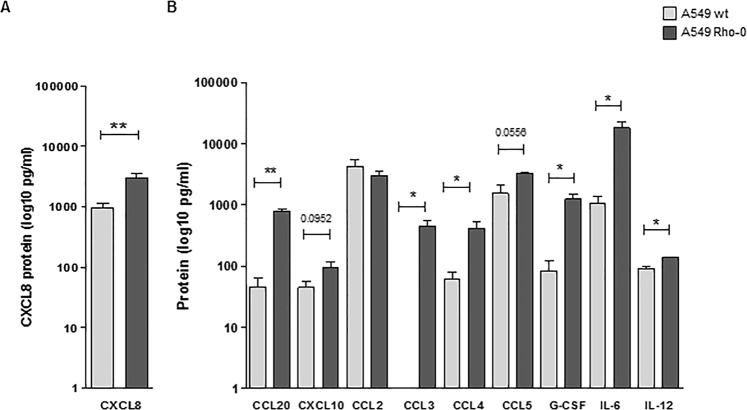
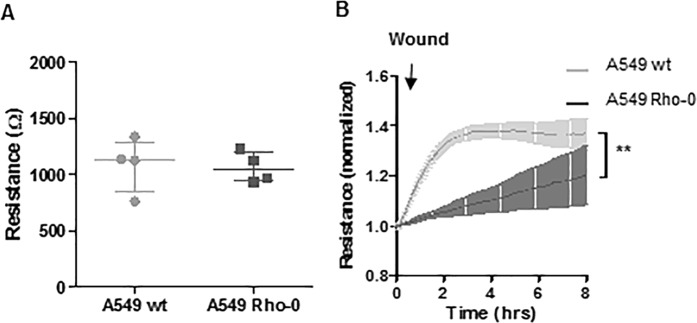
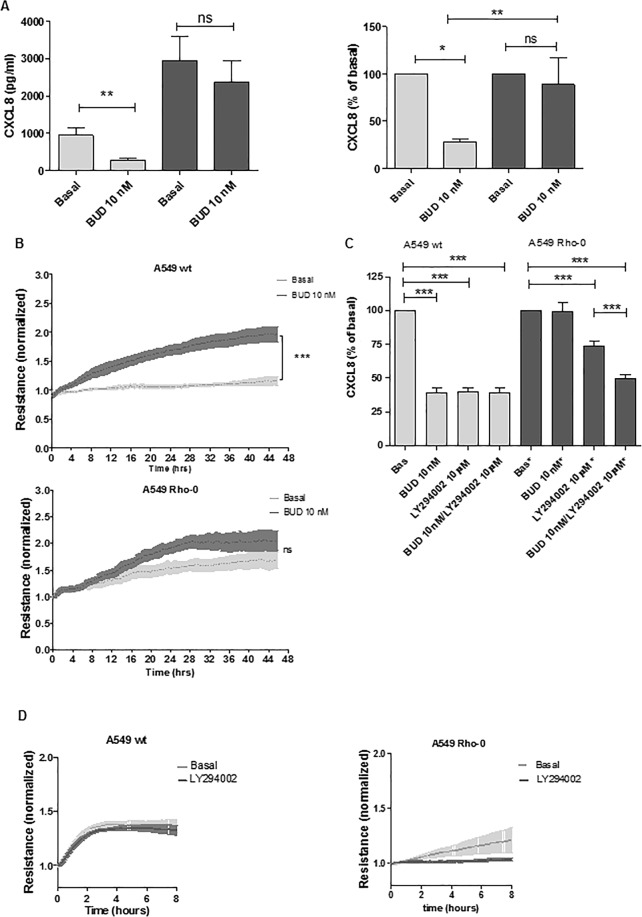
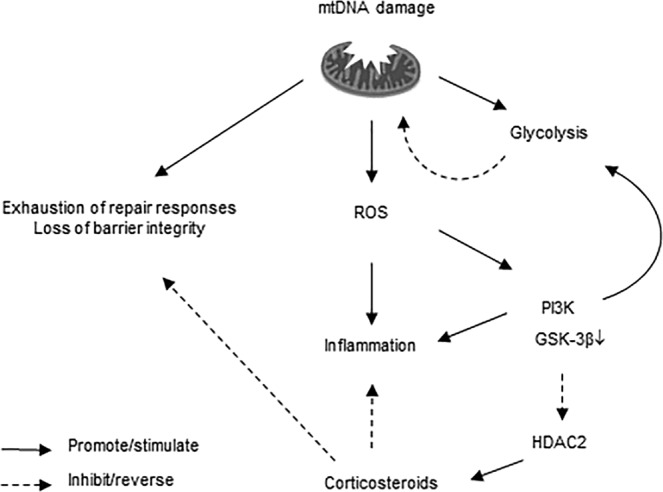
COPD is characterized by chronic lung inflammation and irreversible lung tissue damage. Inhaled noxious gases, including cigarette smoke, are the major risk factor for COPD. Inhaled smoke first encounters the epithelial lining of the lungs, causing oxidative stress and mitochondrial dysfunction. We investigated whether a mitochondrial defect may contribute to increased lung epithelial pro-inflammatory responses, impaired epithelial repair and reduced corticosteroid sensitivity as observed in COPD. We used wild-type alveolar epithelial cells A549 and mitochondrial DNA-depleted A549 cells (A549 Rho-0) and studied pro-inflammatory responses using (multiplex) ELISA as well as epithelial barrier function and repair (real-time impedance measurements), in the presence and absence of the inhaled corticosteroid budesonide. We observed that A549 Rho-0 cells secrete higher levels of pro-inflammatory cytokines than wild-type A549 cells and display impaired repair upon wounding. Budesonide strongly suppressed the production of neutrophil attractant CXCL8, and promoted epithelial integrity in A549 wild-type cells, while A549 Rho-0 cells displayed reduced corticosteroid sensitivity compared to wild-type cells. The reduced corticosteroid responsiveness may be mediated by glycolytic reprogramming, specifically glycolysis-associated PI3K signaling, as PI3K inhibitor LY294002 restored the sensitivity of CXCL8 secretion to corticosteroids in A549 Rho-0 cells. In conclusion, mitochondrial defects may lead to increased lung epithelial pro-inflammatory responses, reduced epithelial repair and reduced corticosteroid responsiveness in lung epithelium, thus potentially contributing to the pathogenesis of COPD.
Conflict of interest statement
This study was funded by and performed within the framework of the Top Institute Pharma project T1-201 “COPD, transition of systemic inflammation into multi-organ pathology”, with partners of the University of Groningen, University Medical Center Groningen, University Medical Center Utrecht, University Medical Center Maastricht, Nycomed BV, GlaxoSmithKline, Danone, AstraZeneca and Foundation TI Pharma. We have no non-financial competing interests.
Figures
References
-
- Heijink, I. et al. Oxidant-induced corticosteroid unresponsiveness in human bronchial epithelial cells. Thorax, 10.1136/thoraxjnl-2013-203520 (2013). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
