

A dimerization-based fluorogenic dye-aptamer module for RNA imaging in live cells
- PMID: 31636432
- PMCID: PMC6920041
- DOI: 10.1038/s41589-019-0381-8
A dimerization-based fluorogenic dye-aptamer module for RNA imaging in live cells
Abstract
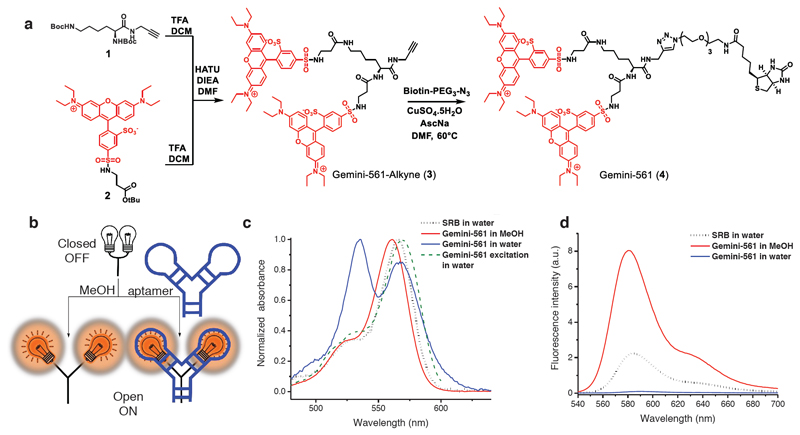
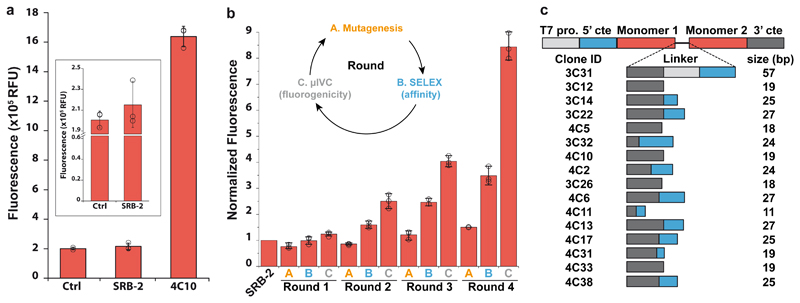
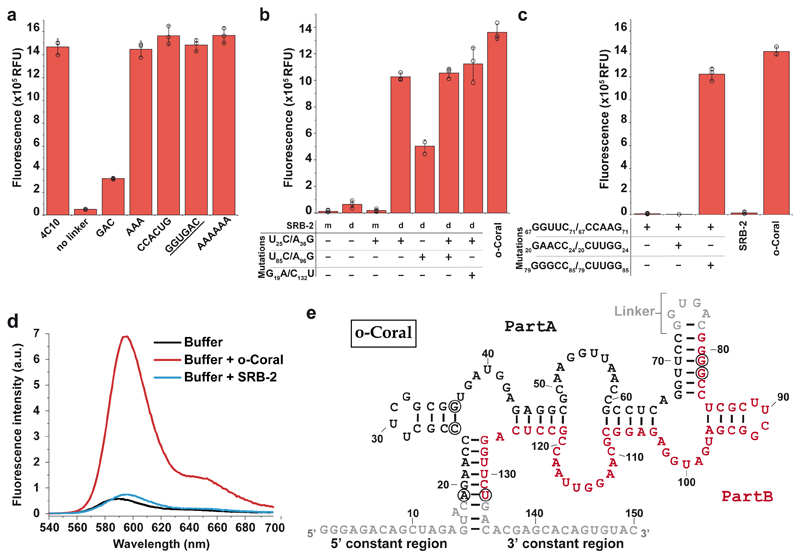
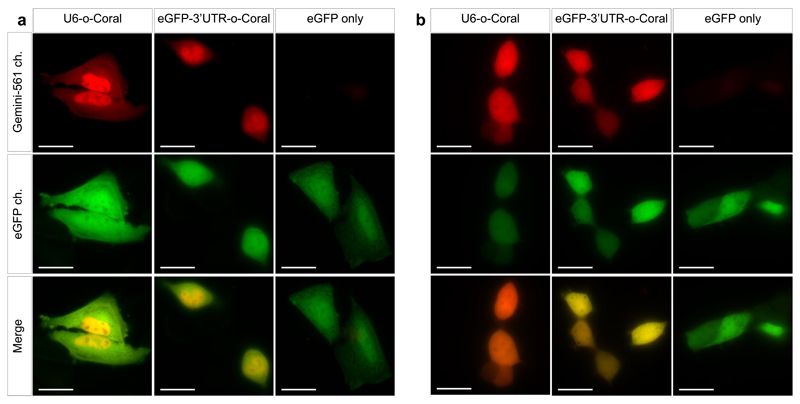
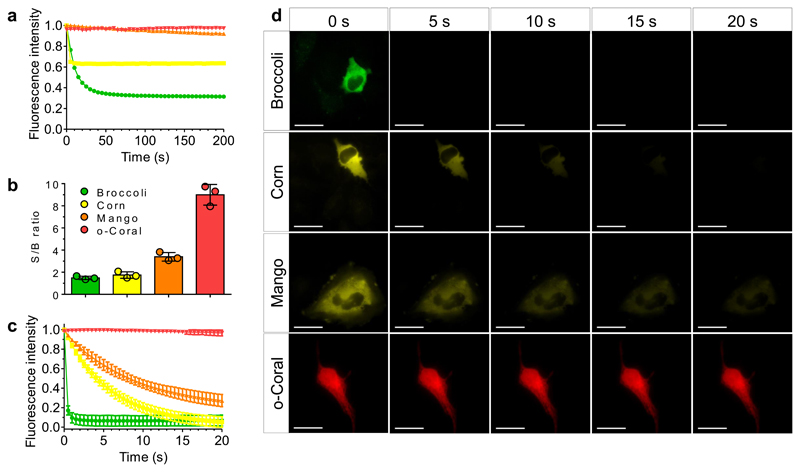
Live-cell imaging of RNA has remained a challenge because of the lack of naturally fluorescent RNAs. Recently developed RNA aptamers that can light-up small fluorogenic dyes could overcome this limitation, but they still suffer from poor brightness and photostability. Here, we propose the concept of a cell-permeable fluorogenic dimer of self-quenched sulforhodamine B dyes (Gemini-561) and the corresponding dimerized aptamer (o-Coral) that can drastically enhance performance of the current RNA imaging method. The improved brightness and photostability, together with high affinity of this complex, allowed direct fluorescence imaging in live mammalian cells of RNA polymerase III transcription products as well as messenger RNAs labeled with a single copy of the aptamer; that is, without tag multimerization. The developed fluorogenic module enables fast and sensitive detection of RNA inside live cells, while the proposed design concept opens the route to new generation of ultrabright RNA probes.
Conflict of interest statement
F.B., K.T.F., M.C., A.K., M.R., the University of Strasbourg and the CNRS have filed a patent application covering the technology presented in this manuscript.
Figures
References
-
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–24. - PubMed
-
- Xue L, Karpenko IA, Hiblot J, Johnsson K. Imaging and manipulating proteins in live cells through covalent labeling. Nat Chem Biol. 2015;11:917–23. - PubMed
-
- Bertrand E, et al. Localization of ASH1 mRNA particles in living yeast. Mol Cell. 1998;2:437–45. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
