

History and progress of hypotheses and clinical trials for Alzheimer's disease
- PMID: 31637009
- PMCID: PMC6799833
- DOI: 10.1038/s41392-019-0063-8
History and progress of hypotheses and clinical trials for Alzheimer's disease
Erratum in
-
Erratum: Author Correction: History and progress of hypotheses and clinical trials for Alzheimer's disease.Signal Transduct Target Ther. 2019 Sep 23;4:37. doi: 10.1038/s41392-019-0071-8. eCollection 2019. Signal Transduct Target Ther. 2019. PMID: 31638618 Free PMC article.
Abstract
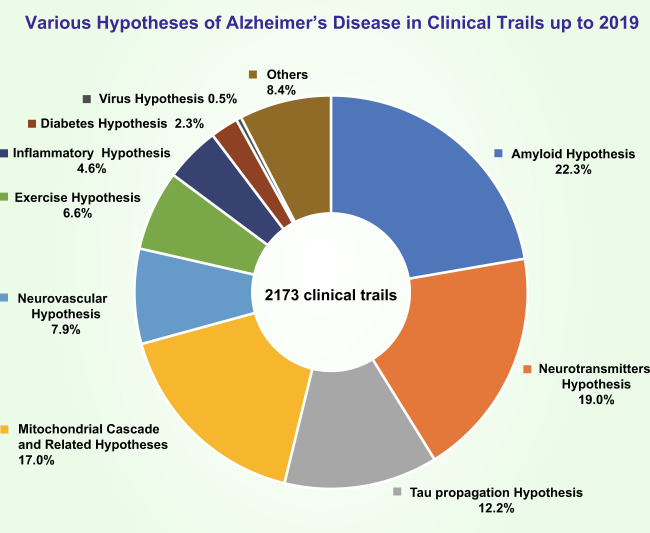
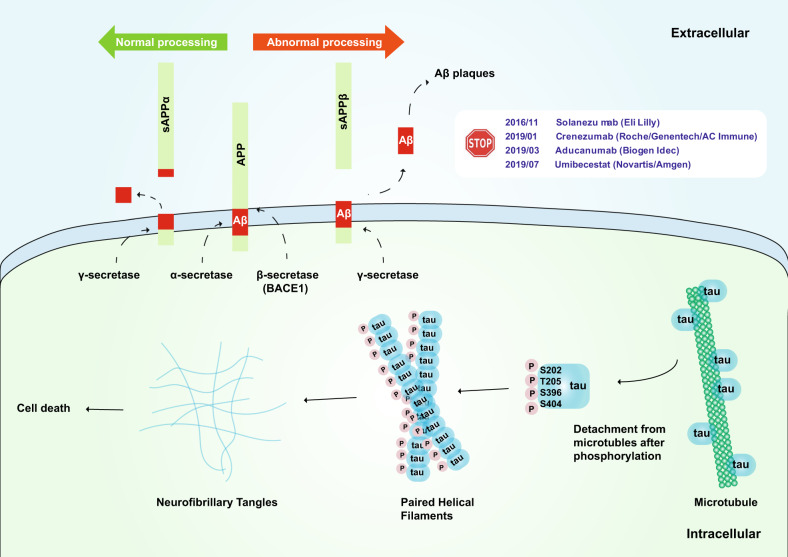
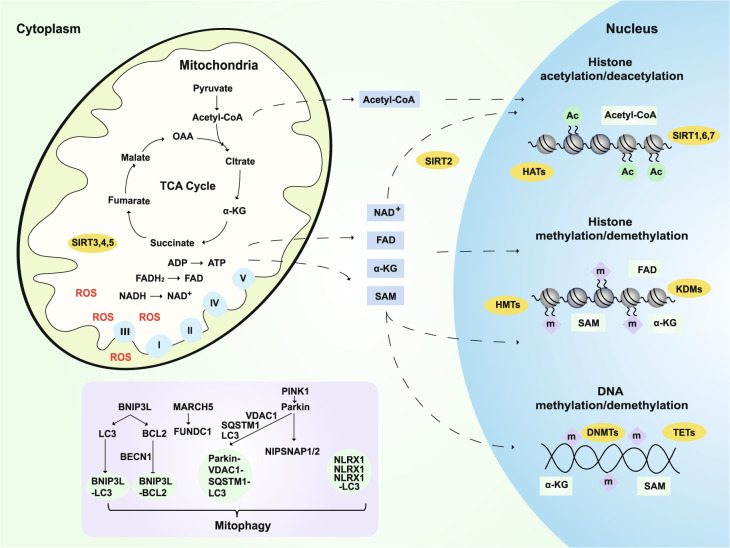
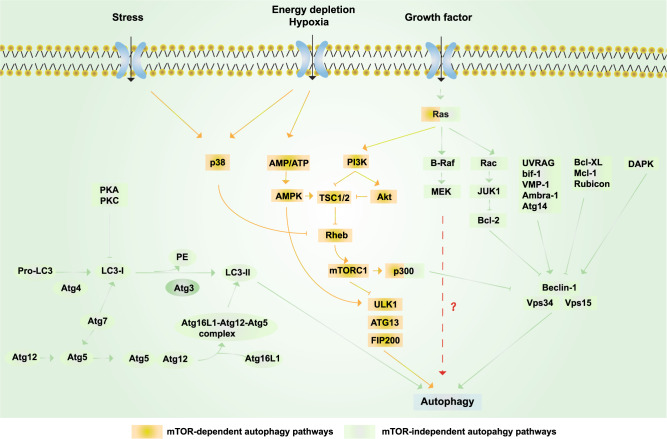
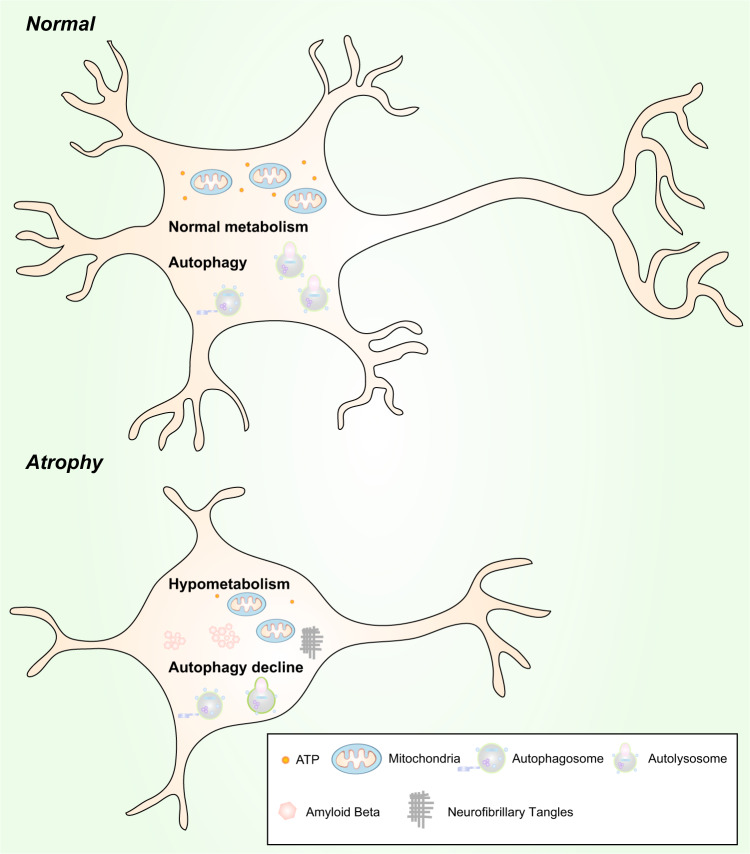
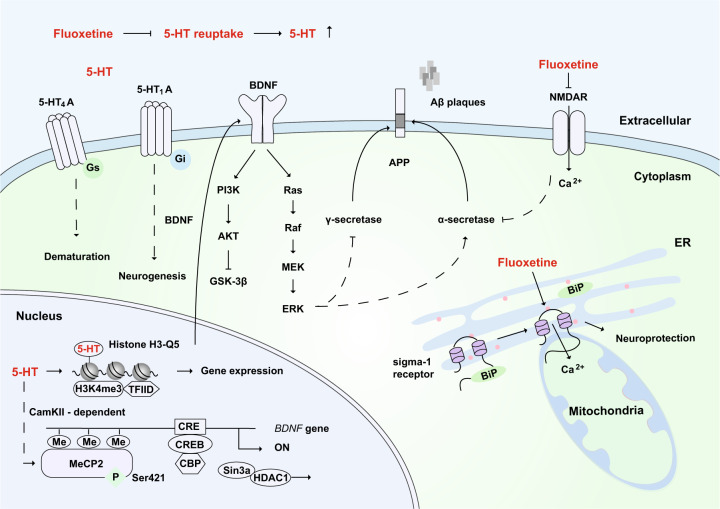
Alzheimer's disease (AD) is a neurodegenerative disease characterized by progressive memory loss along with neuropsychiatric symptoms and a decline in activities of daily life. Its main pathological features are cerebral atrophy, amyloid plaques, and neurofibrillary tangles in the brains of patients. There are various descriptive hypotheses regarding the causes of AD, including the cholinergic hypothesis, amyloid hypothesis, tau propagation hypothesis, mitochondrial cascade hypothesis, calcium homeostasis hypothesis, neurovascular hypothesis, inflammatory hypothesis, metal ion hypothesis, and lymphatic system hypothesis. However, the ultimate etiology of AD remains obscure. In this review, we discuss the main hypotheses of AD and related clinical trials. Wealthy puzzles and lessons have made it possible to develop explanatory theories and identify potential strategies for therapeutic interventions for AD. The combination of hypometabolism and autophagy deficiency is likely to be a causative factor for AD. We further propose that fluoxetine, a selective serotonin reuptake inhibitor, has the potential to treat AD.
Keywords: Neurological disorders; Neuroscience.
© The Author(s) 2019.
Conflict of interest statement
Competing interestsThe authors declare no competing interests.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
