

Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo
- PMID: 31639368
- PMCID: PMC6838682
- DOI: 10.1016/j.devcel.2019.10.003
Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo
Abstract
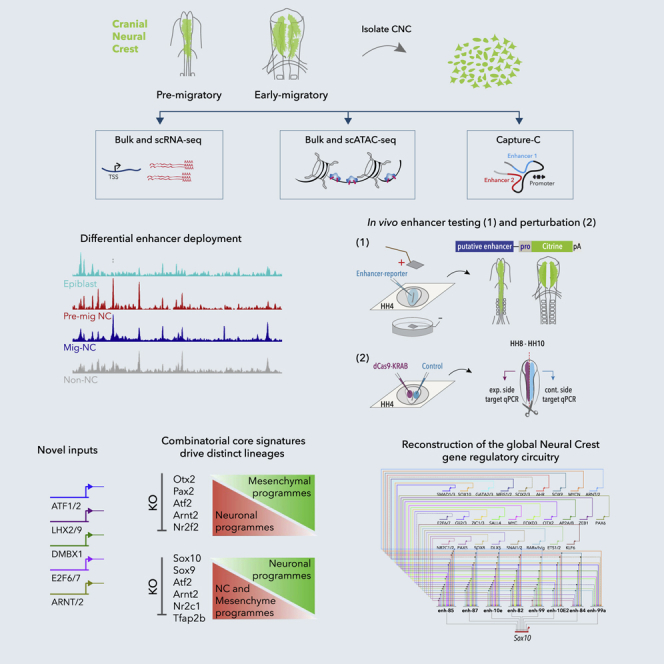
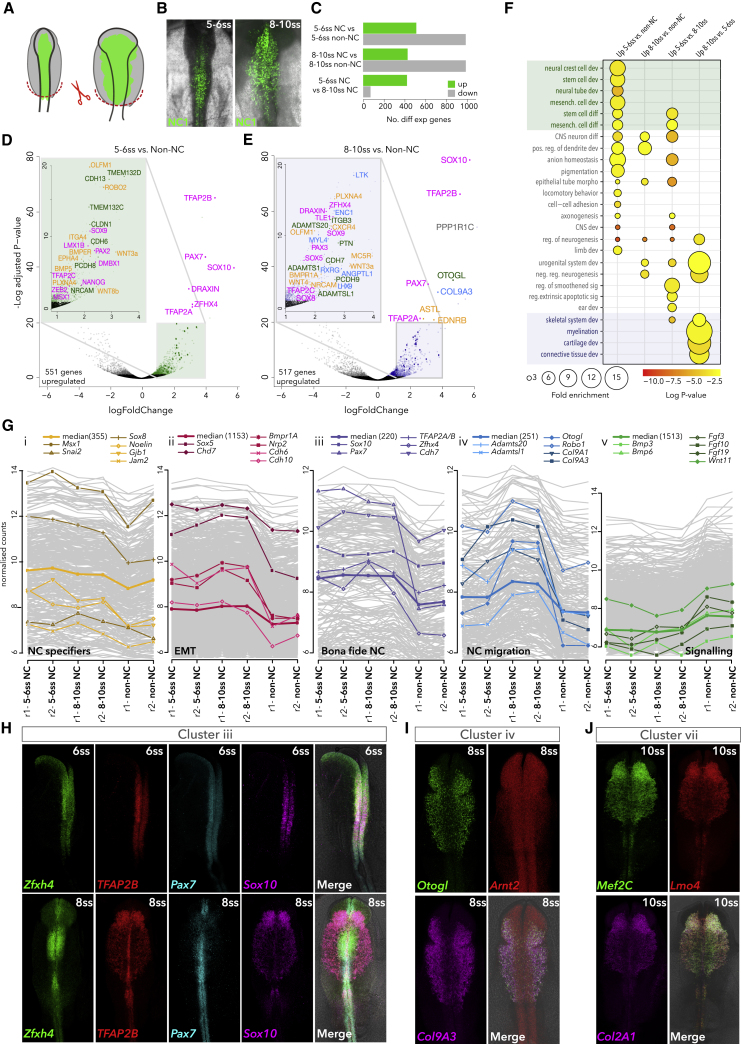
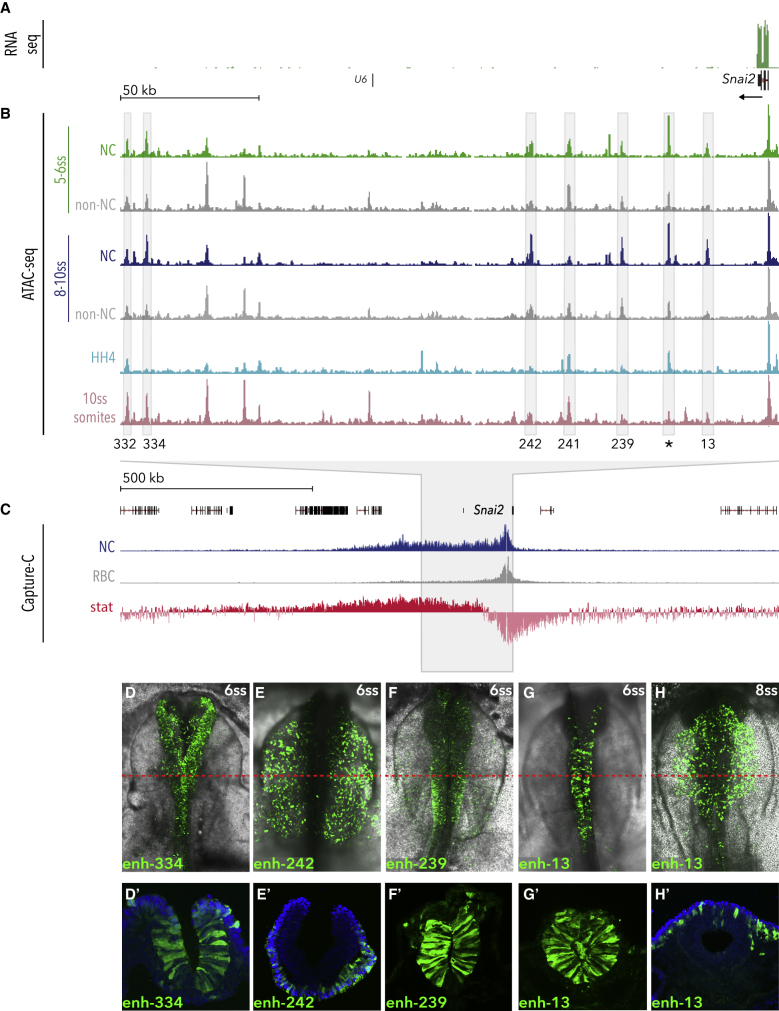
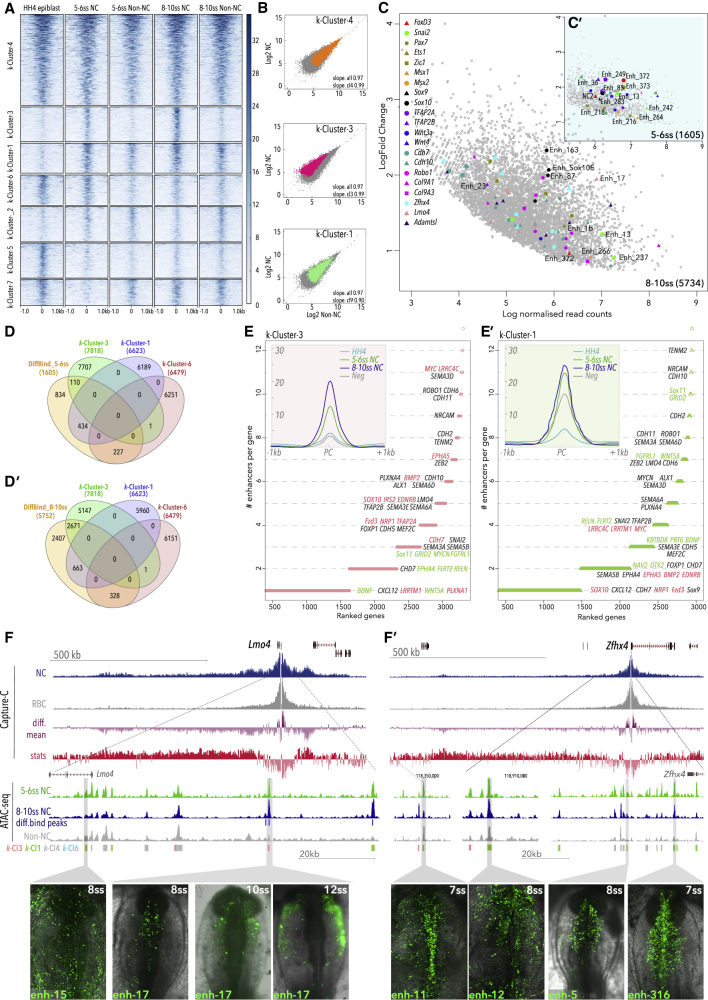
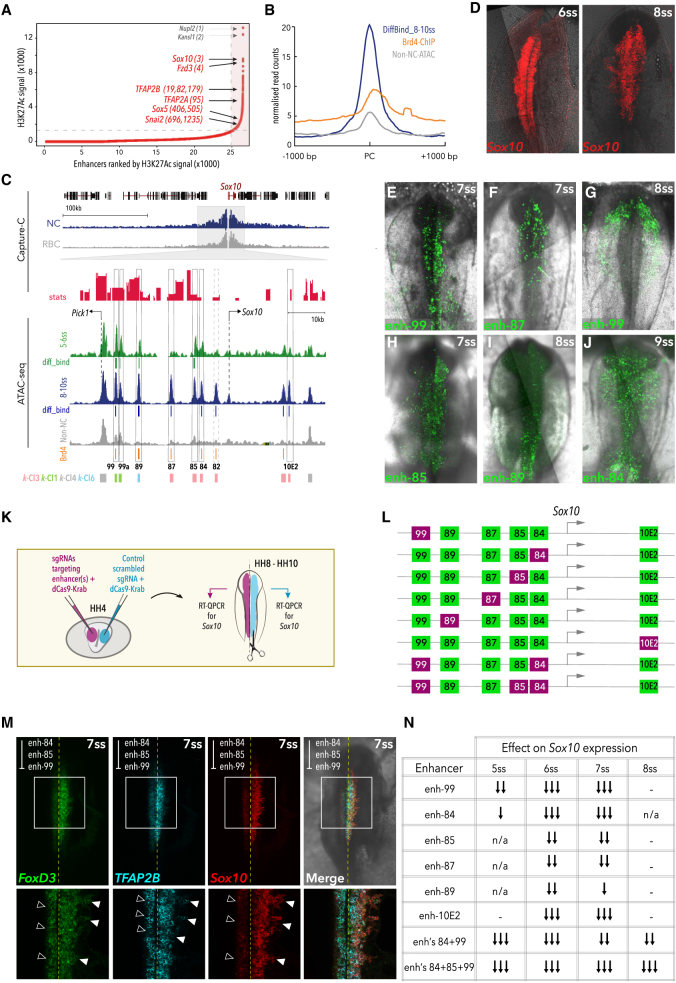
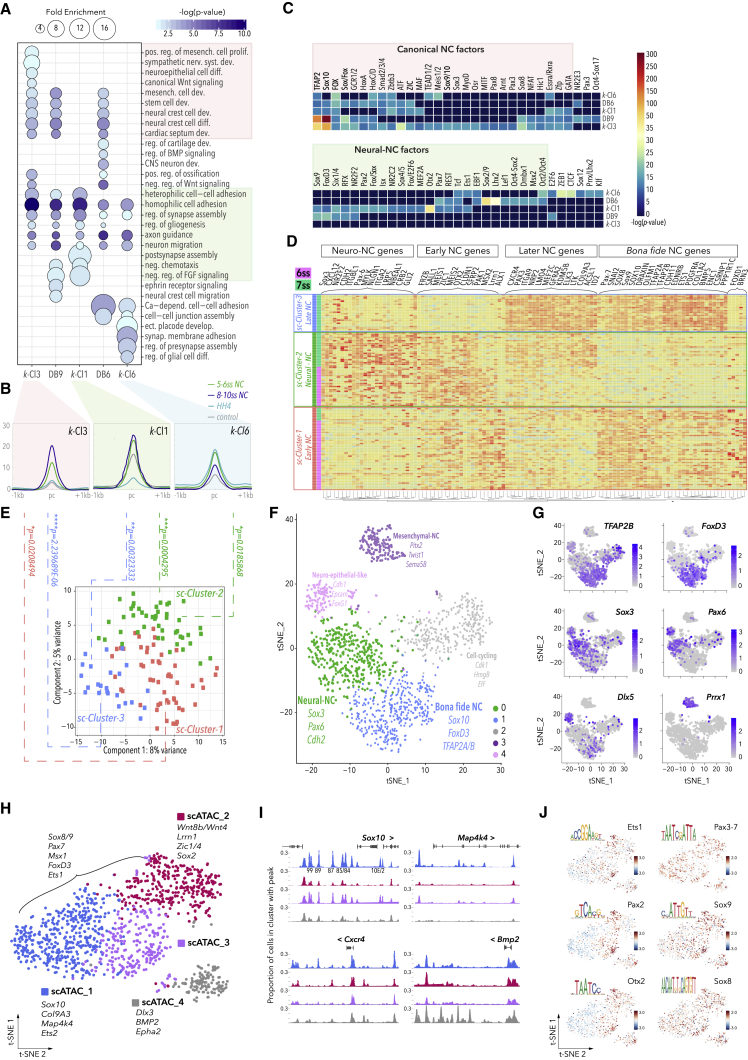
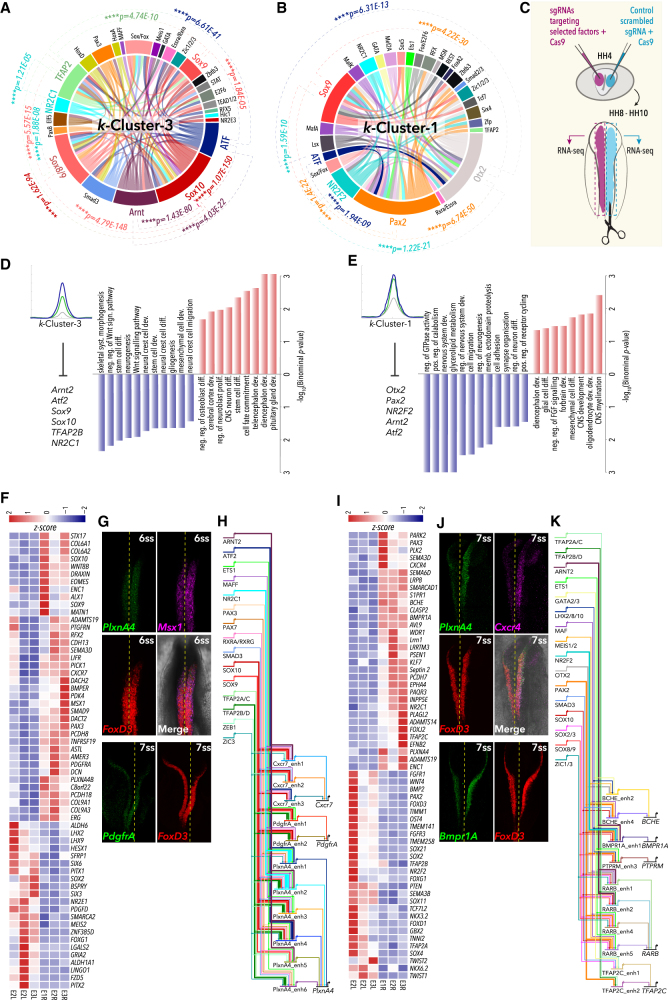
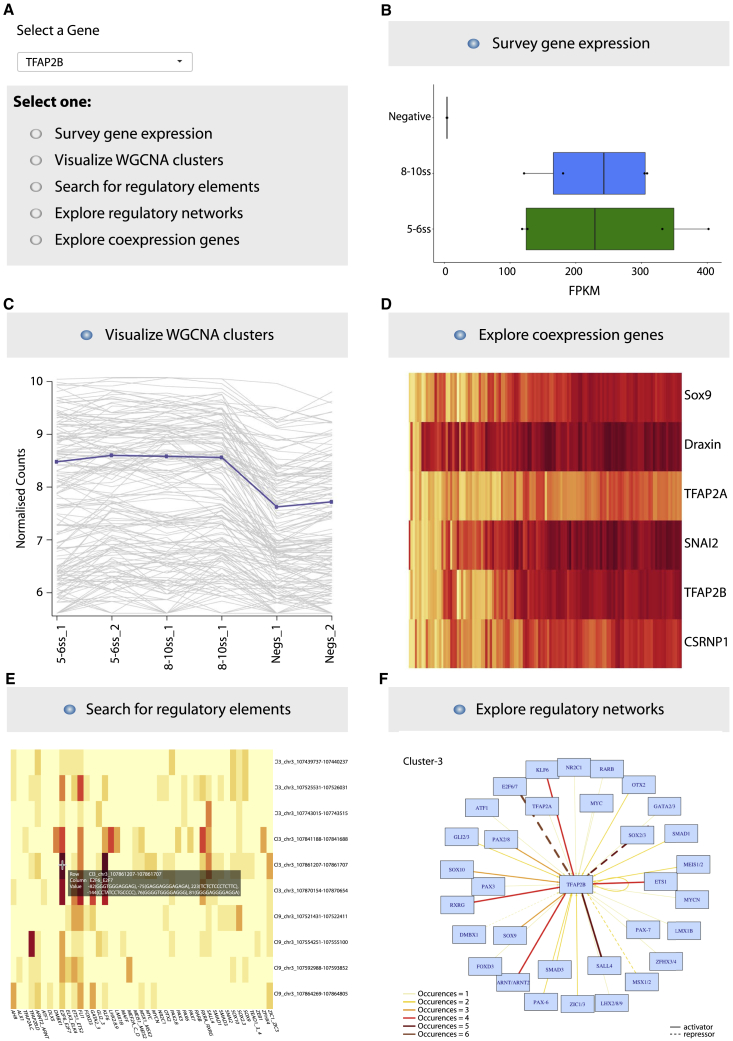
Precise control of developmental processes is encoded in the genome in the form of gene regulatory networks (GRNs). Such multi-factorial systems are difficult to decode in vertebrates owing to their complex gene hierarchies and dynamic molecular interactions. Here we present a genome-wide in vivo reconstruction of the GRN underlying development of the multipotent neural crest (NC) embryonic cell population. By coupling NC-specific epigenomic and transcriptional profiling at population and single-cell levels with genome/epigenome engineering in vivo, we identify multiple regulatory layers governing NC ontogeny, including NC-specific enhancers and super-enhancers, novel trans-factors, and cis-signatures allowing reverse engineering of the NC-GRN at unprecedented resolution. Furthermore, identification and dissection of divergent upstream combinatorial regulatory codes has afforded new insights into opposing gene circuits that define canonical and neural NC fates early during NC ontogeny. Our integrated approach, allowing dissection of cell-type-specific regulatory circuits in vivo, has broad implications for GRN discovery and investigation.
Keywords: ATAC; Capture-C; chick; enhancers; gene regulatory network; neural crest; scATAC-seq; super-enhancers; transcription factors, scRNA-seq.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Acampora D., Mazan S., Lallemand Y., Avantaggiato V., Maury M., Simeone A., Brûlet P. Forebrain and midbrain regions are deleted in Otx2-/- mutants due to a defective anterior neuroectoderm specification during gastrulation. Development. 1995;121:3279–3290. - PubMed
-
- Aguiar D.P., Sghari S., Creuzet S. The facial neural crest controls fore- and midbrain patterning by regulating Foxg1 expression through Smad1 activity. Development. 2014;141:2494–2505. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
