

Exploring a Pool-seq-only approach for gaining population genomic insights in nonmodel species
- PMID: 31641485
- PMCID: PMC6802065
- DOI: 10.1002/ece3.5646
Exploring a Pool-seq-only approach for gaining population genomic insights in nonmodel species
Abstract
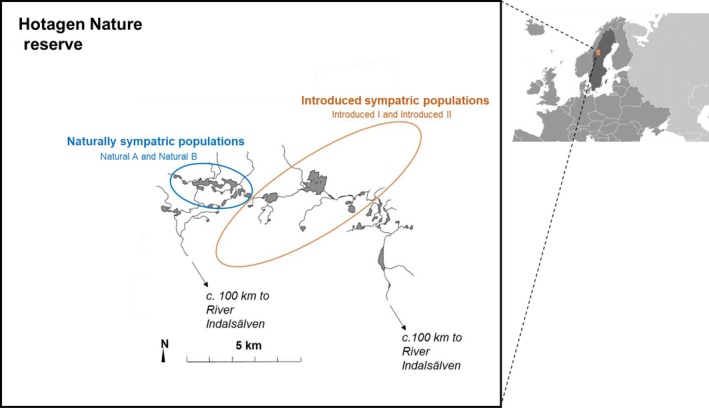
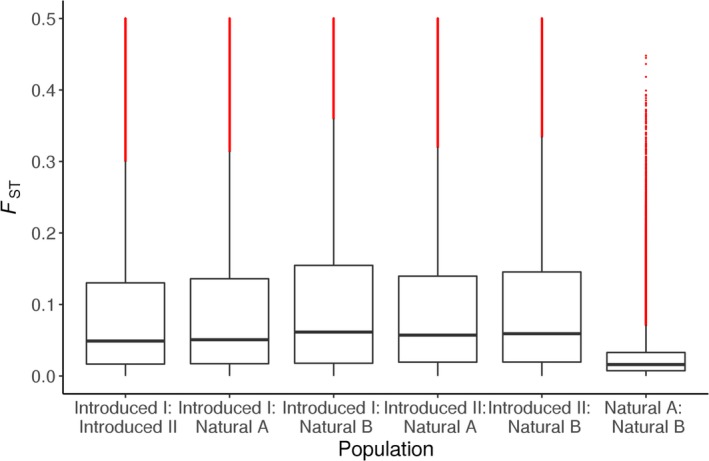
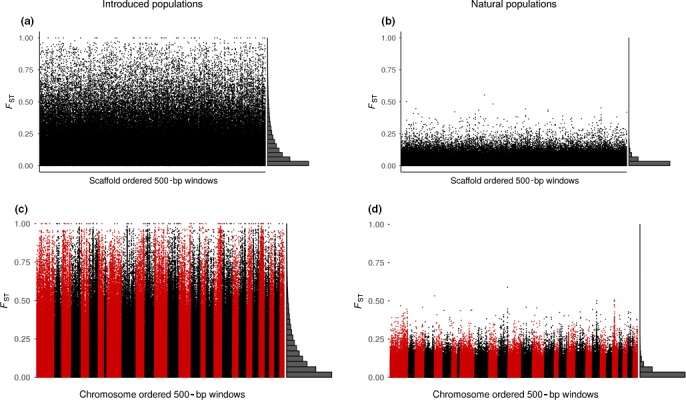
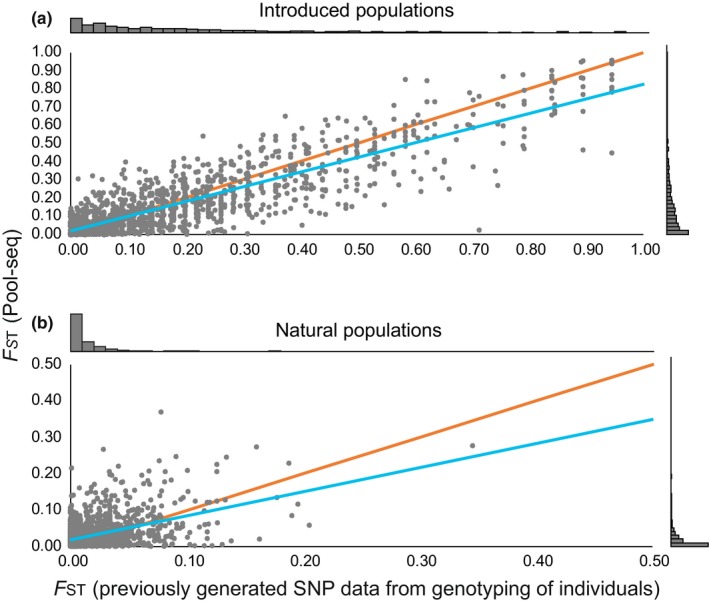
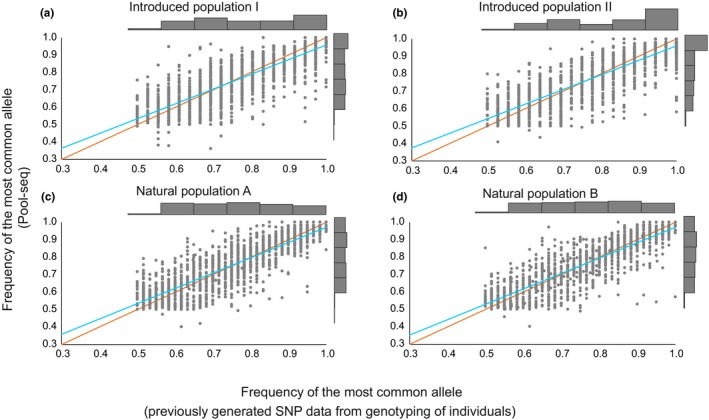
Developing genomic insights is challenging in nonmodel species for which resources are often scarce and prohibitively costly. Here, we explore the potential of a recently established approach using Pool-seq data to generate a de novo genome assembly for mining exons, upon which Pool-seq data are used to estimate population divergence and diversity. We do this for two pairs of sympatric populations of brown trout (Salmo trutta): one naturally sympatric set of populations and another pair of populations introduced to a common environment. We validate our approach by comparing the results to those from markers previously used to describe the populations (allozymes and individual-based single nucleotide polymorphisms [SNPs]) and from mapping the Pool-seq data to a reference genome of the closely related Atlantic salmon (Salmo salar). We find that genomic differentiation (F ST) between the two introduced populations exceeds that of the naturally sympatric populations (F ST = 0.13 and 0.03 between the introduced and the naturally sympatric populations, respectively), in concordance with estimates from the previously used SNPs. The same level of population divergence is found for the two genome assemblies, but estimates of average nucleotide diversity differ ( ≈ 0.002 and ≈ 0.001 when mapping to S. trutta and S. salar, respectively), although the relationships between population values are largely consistent. This discrepancy might be attributed to biases when mapping to a haploid condensed assembly made of highly fragmented read data compared to using a high-quality reference assembly from a divergent species. We conclude that the Pool-seq-only approach can be suitable for detecting and quantifying genome-wide population differentiation, and for comparing genomic diversity in populations of nonmodel species where reference genomes are lacking.
Keywords: Salmo trutta; genetic diversity; genome sequencing; population genomics; salmonid; single nucleotide polymorphism.
© 2019 The Authors. Ecology and Evolution published by John Wiley & Sons Ltd.
Conflict of interest statement
None declared.
Figures

References
-
- Allendorf, F. W. , & Ryman, N. (2002). The role of genetics in population viability analysis In Beissinger S. R. & McCullough D. R. (Eds.), Population viability analysis (pp. 50–85). Chicago, IL: University of Chicago Press.
-
- Andersson, A. , Jansson, E. , Wennerström, L. , Chiriboga, F. , Arnyasi, M. , Kent, M. P. , & Laikre, L. (2017). Complex genetic diversity patterns of cryptic, sympatric brown trout (Salmo trutta) populations in tiny mountain lakes. Conservation Genetics, 18, 1213–1227. 10.1007/s10592-017-0972-4 - DOI
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
