

Somatic mutations and clonal dynamics in healthy and cirrhotic human liver
- PMID: 31645727
- PMCID: PMC6837891
- DOI: 10.1038/s41586-019-1670-9
Somatic mutations and clonal dynamics in healthy and cirrhotic human liver
Abstract
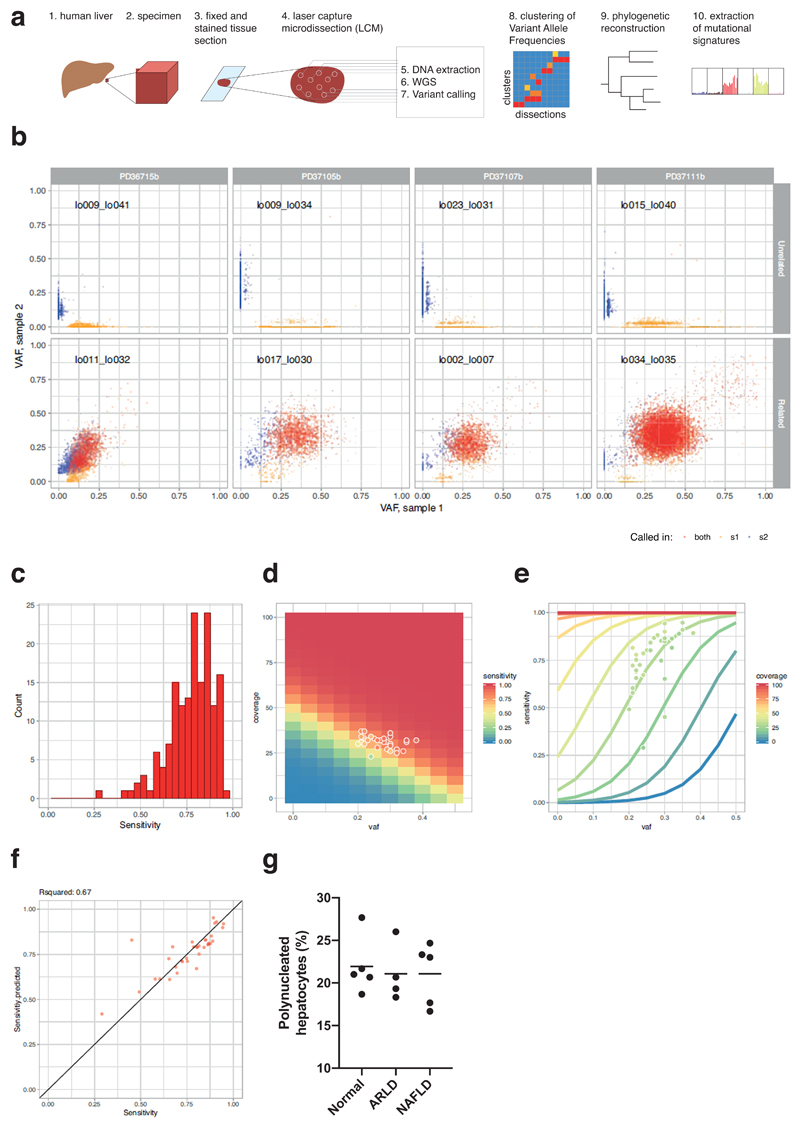
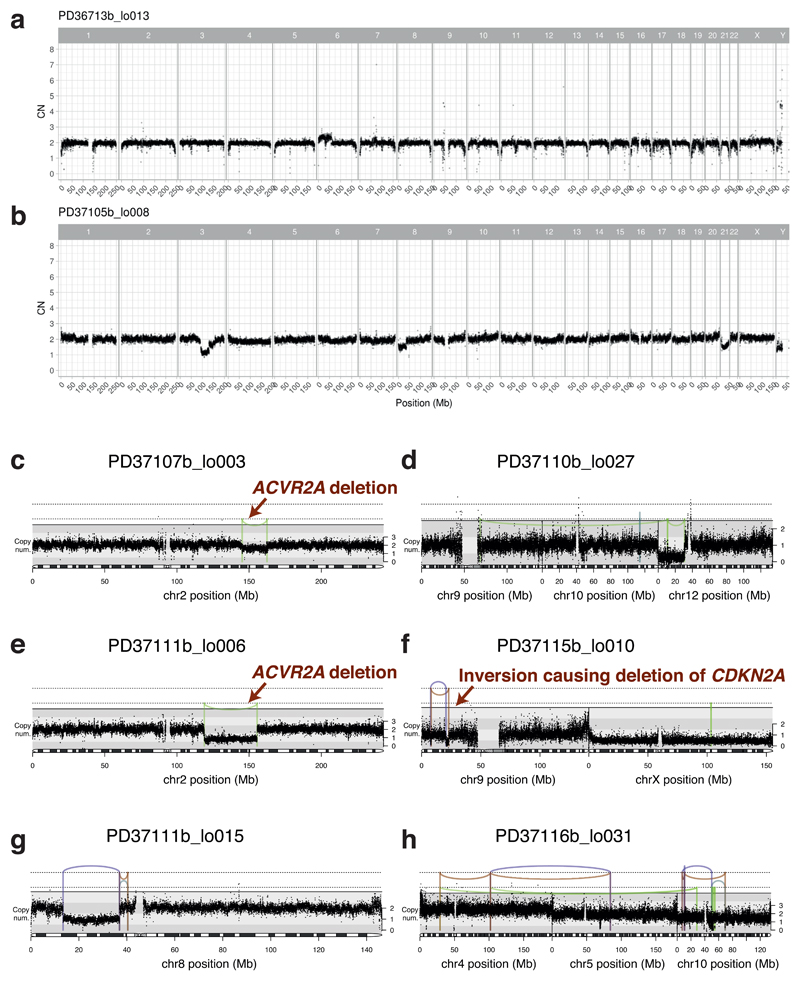
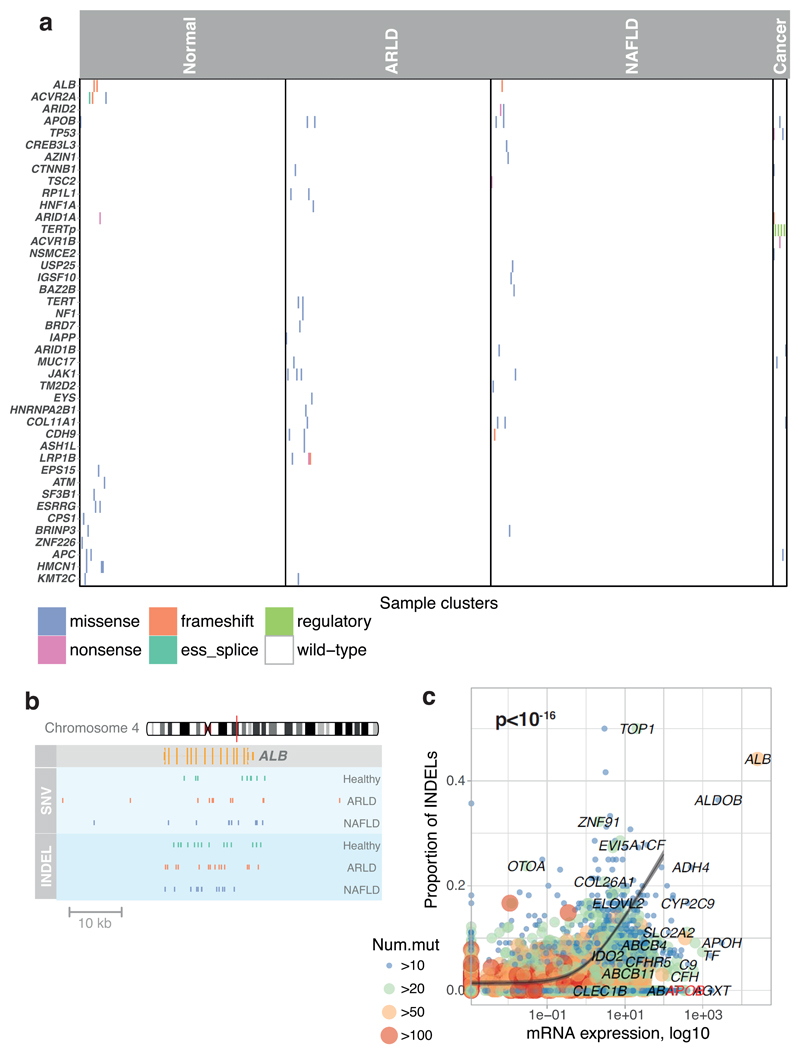
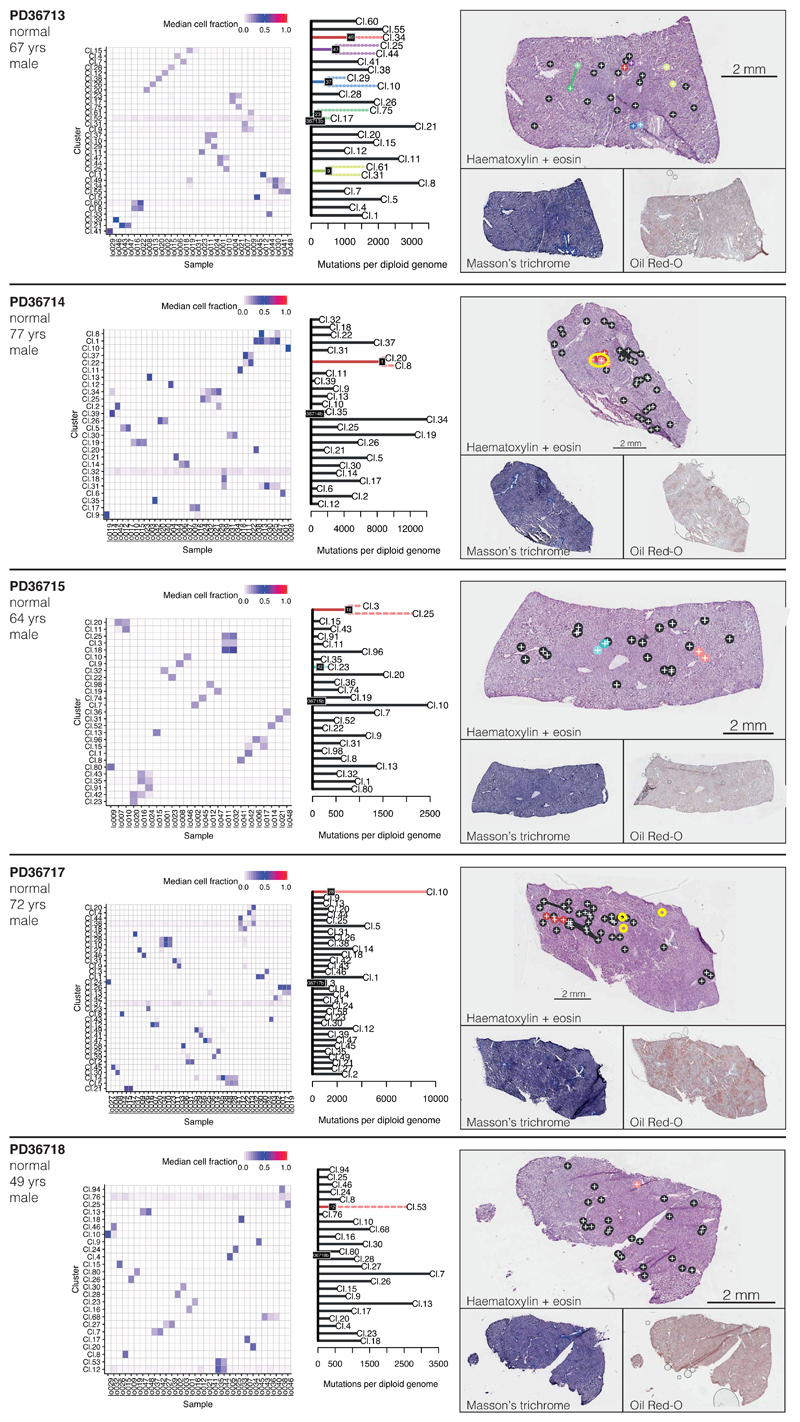
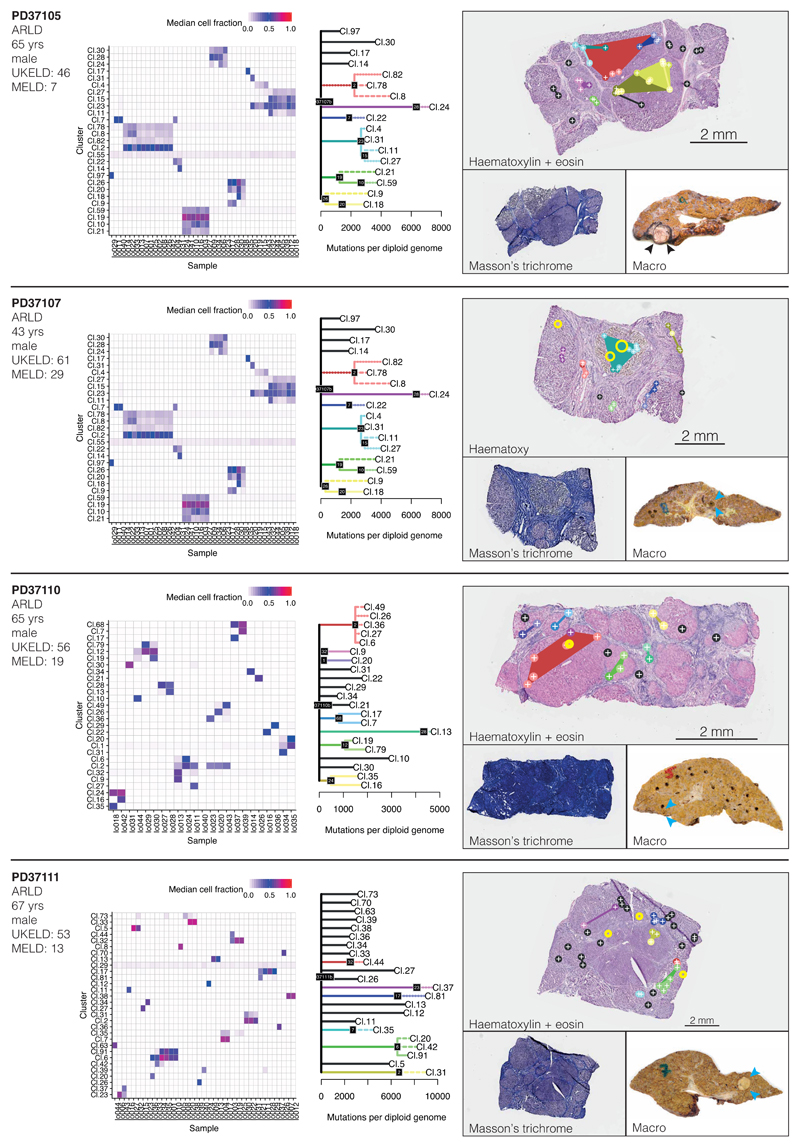
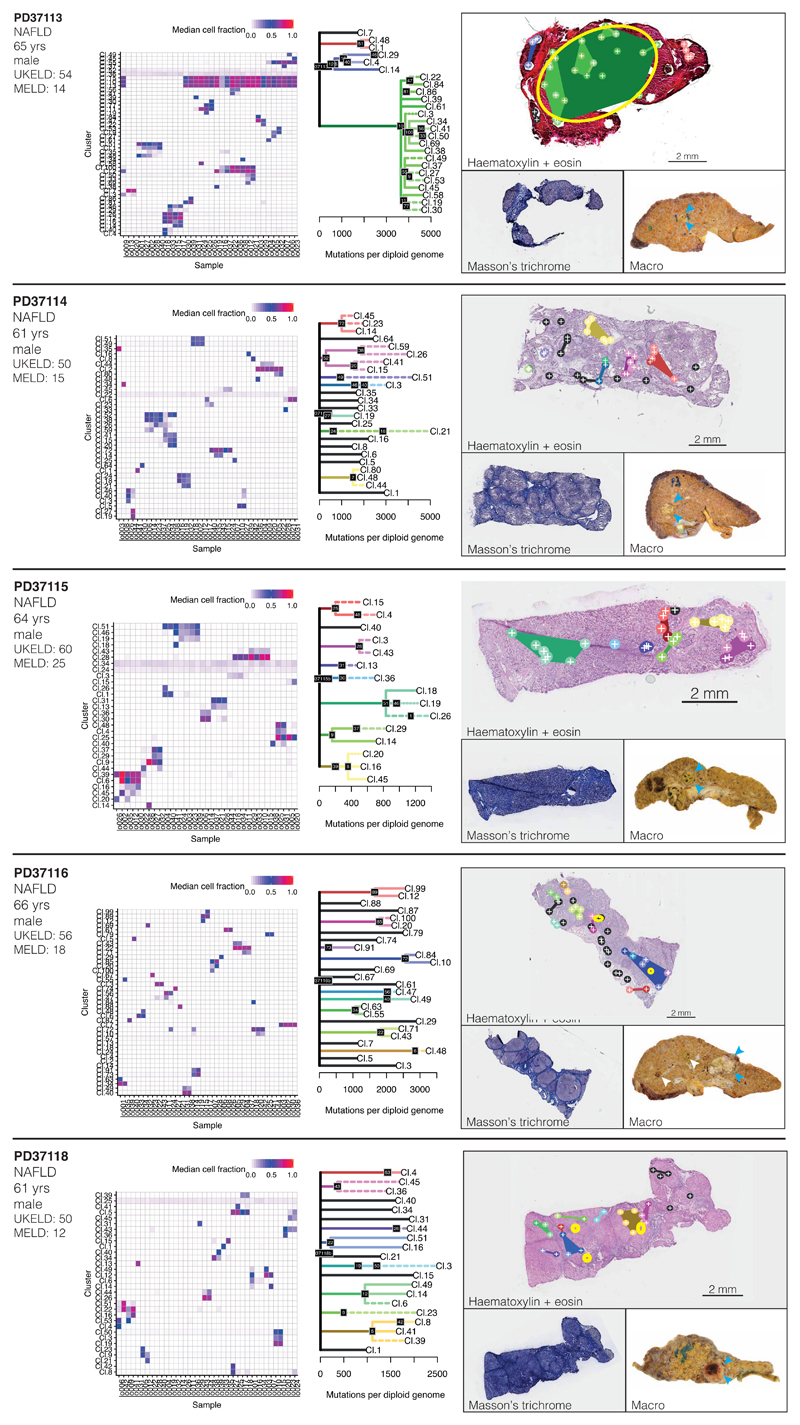
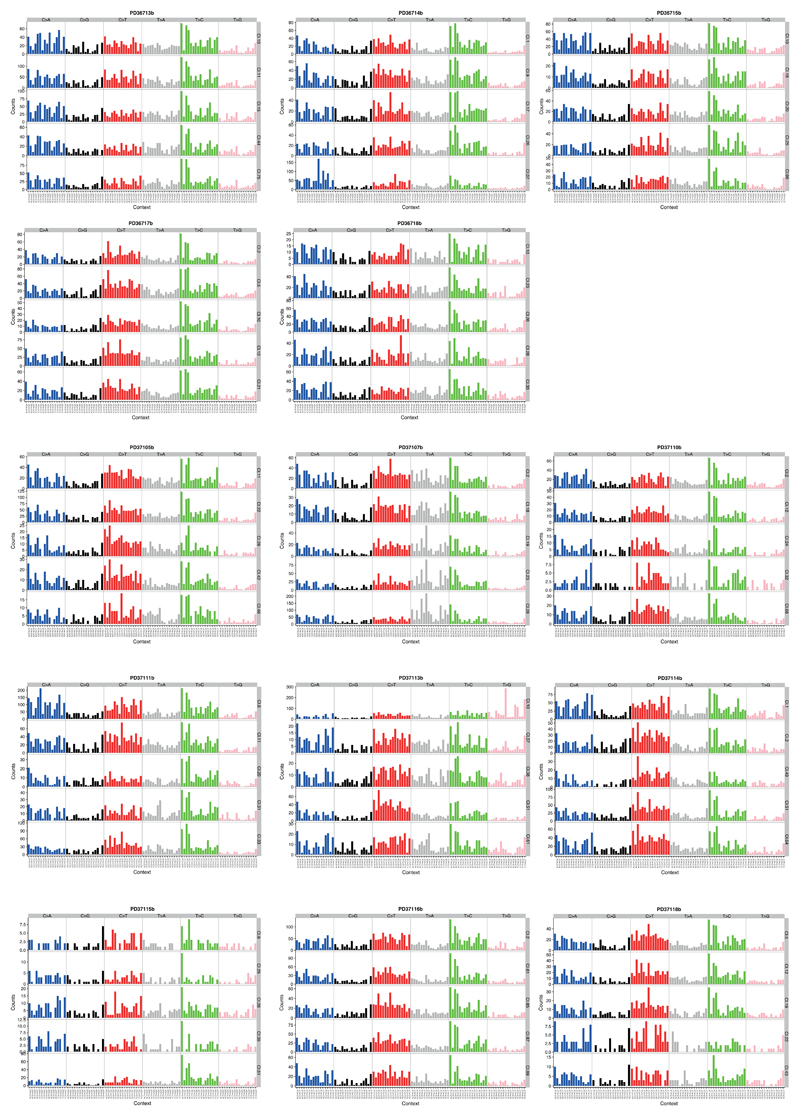
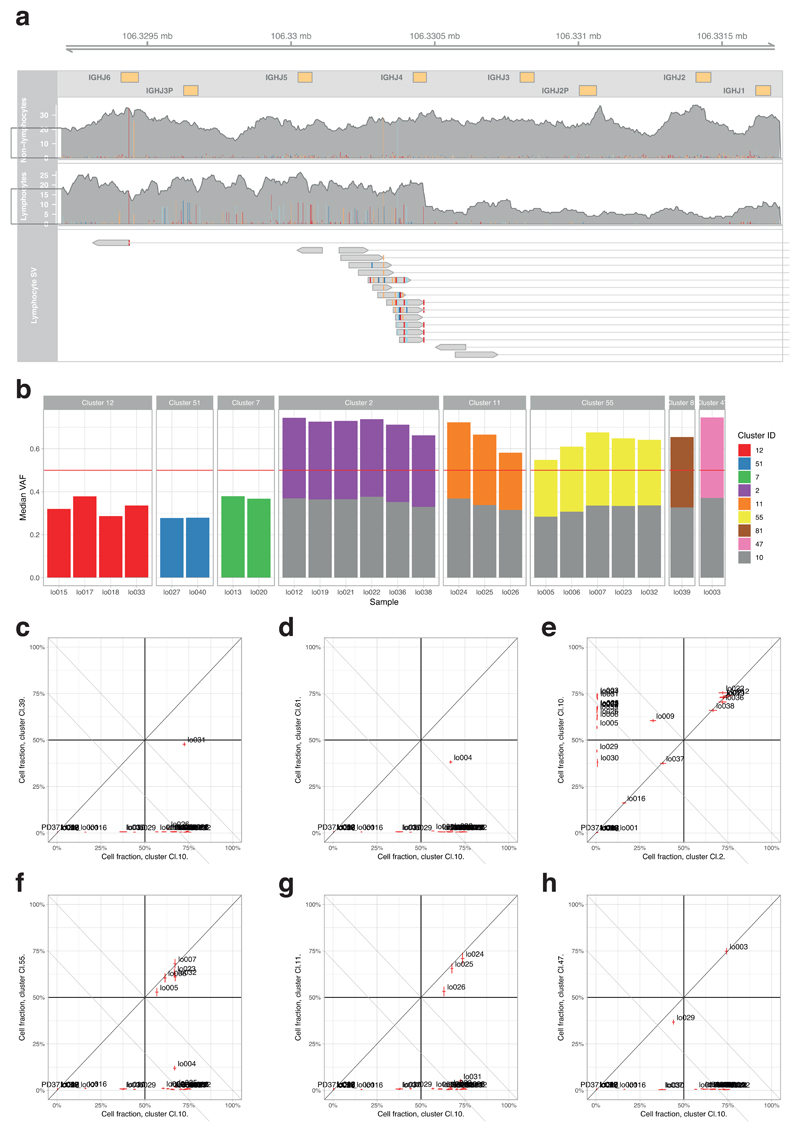
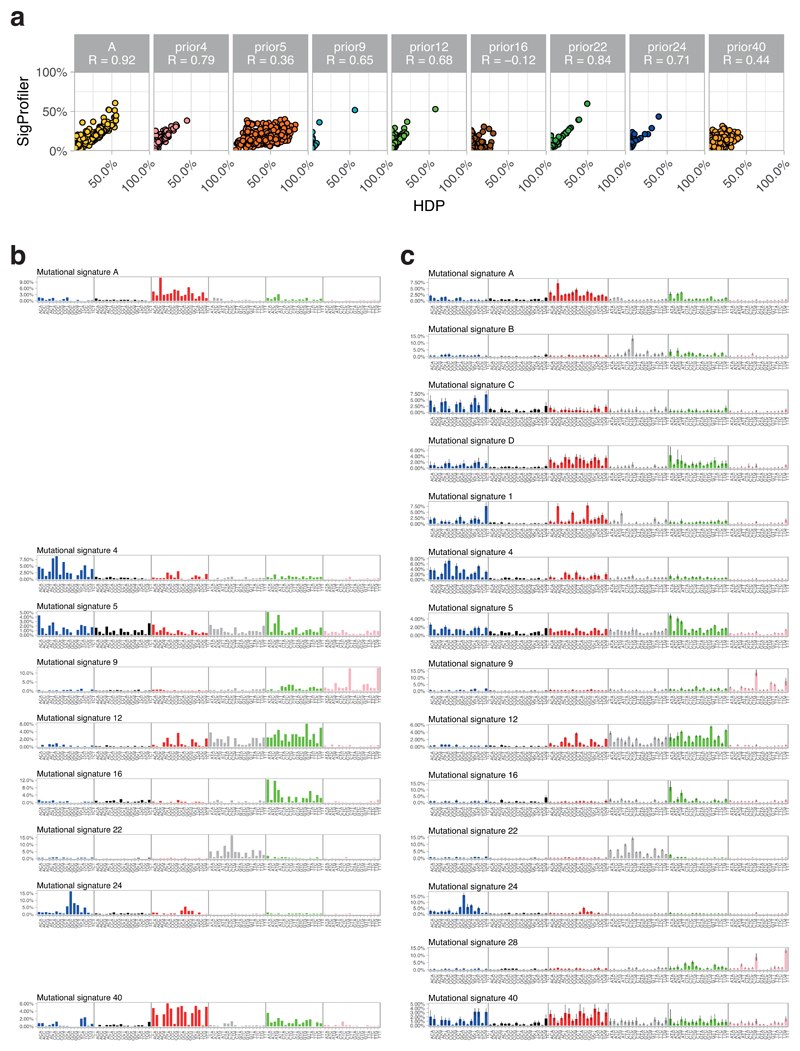
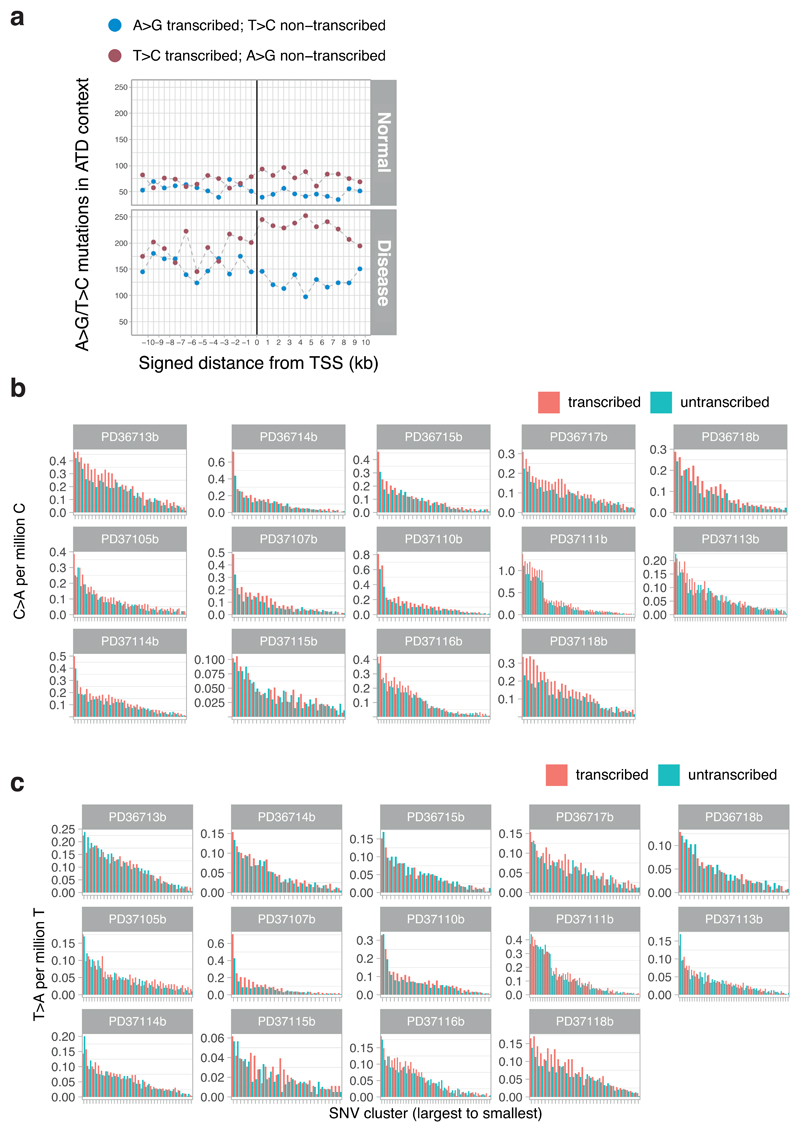
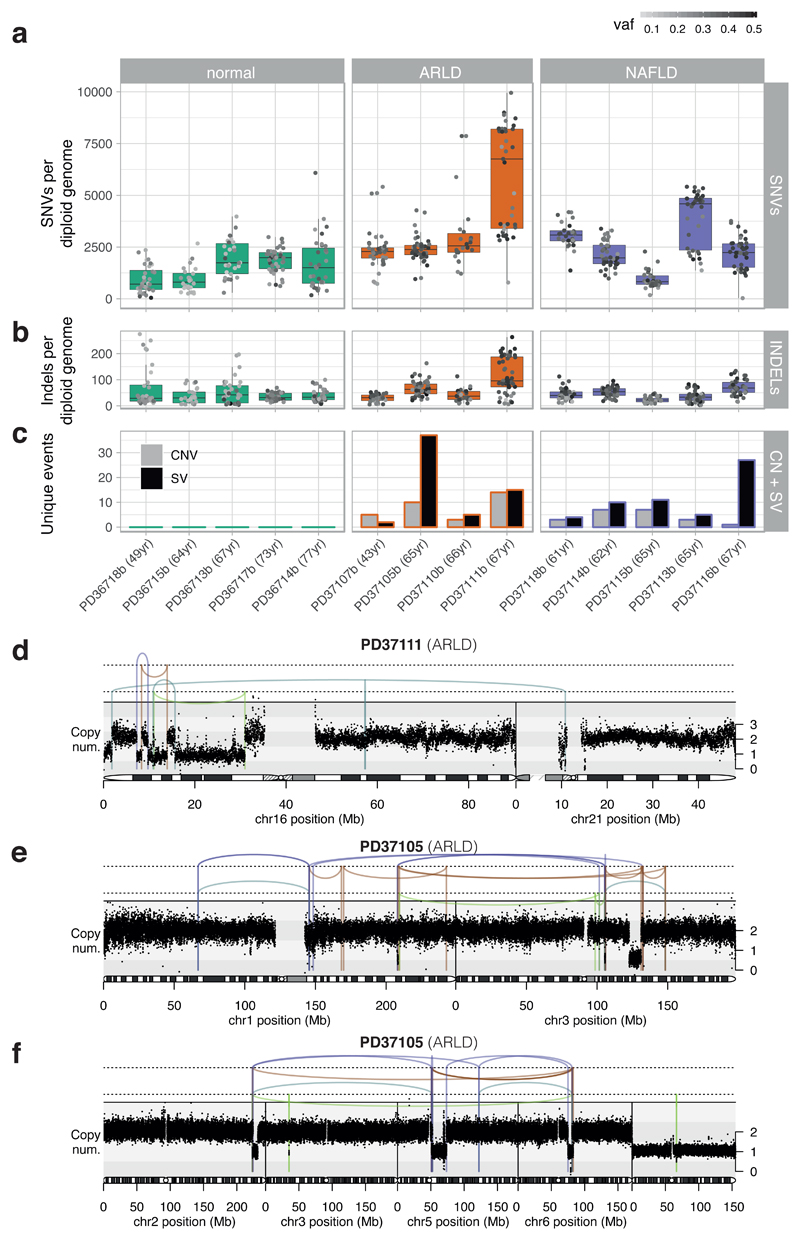
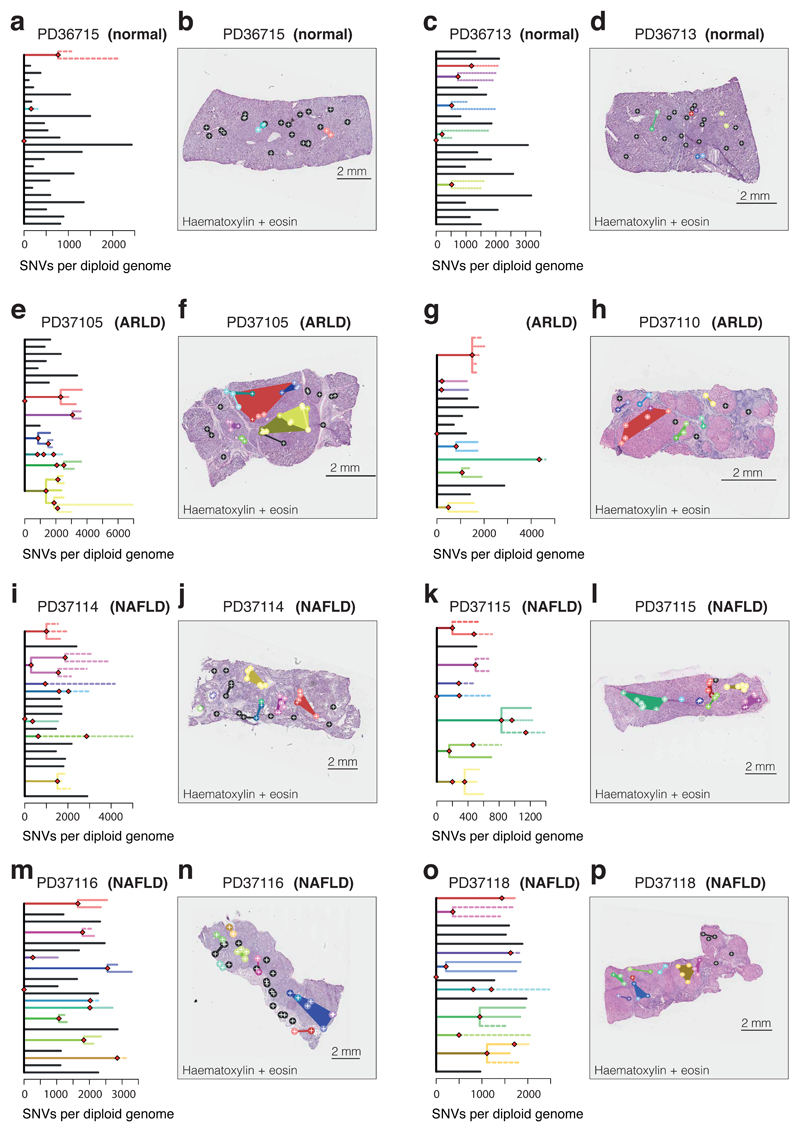
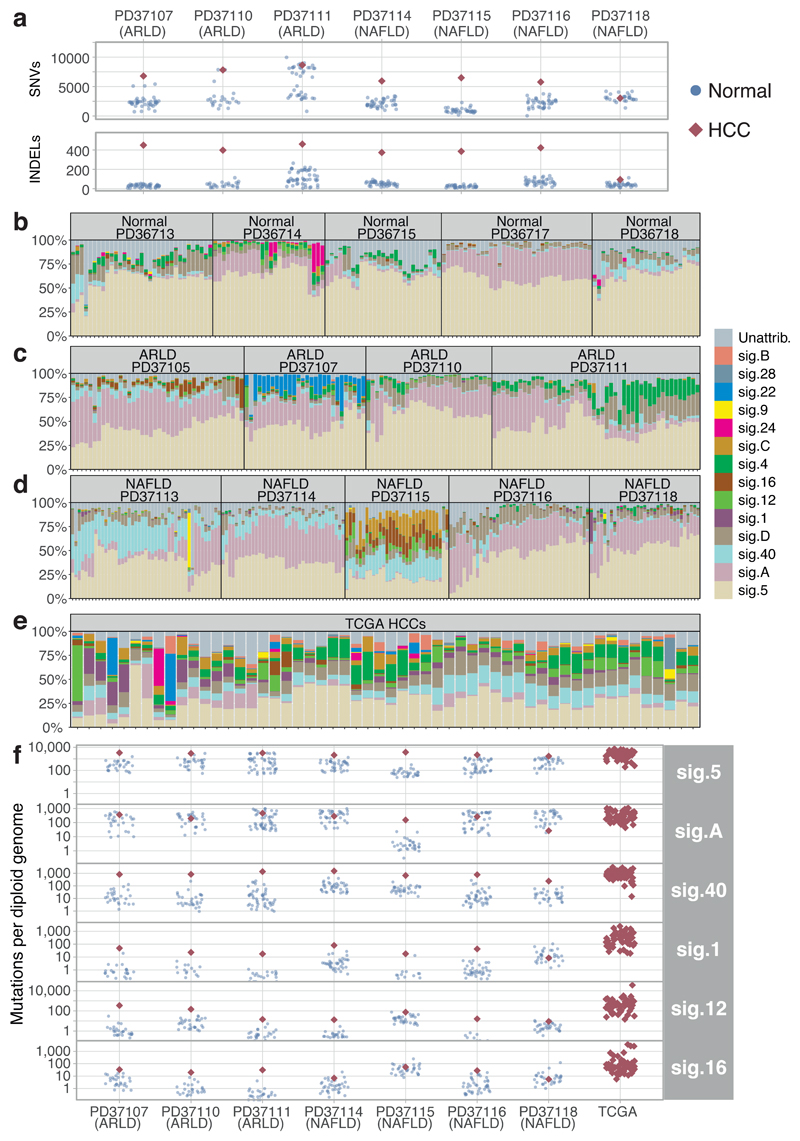
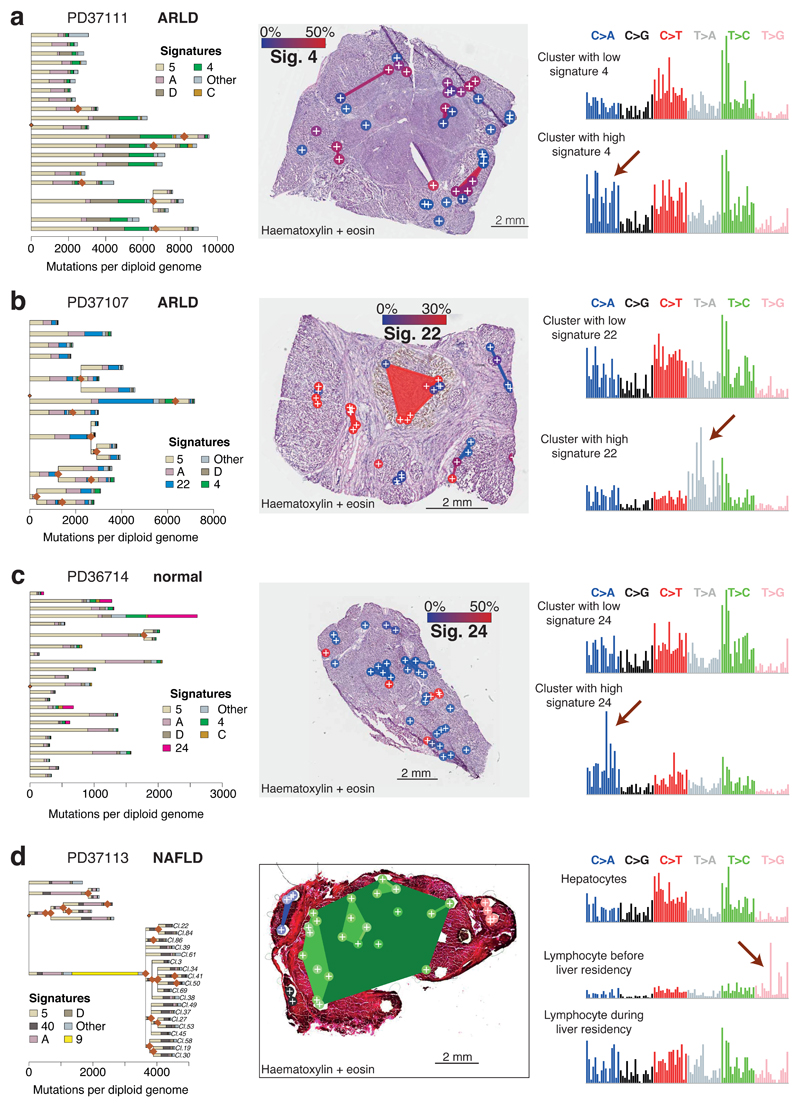
The most common causes of chronic liver disease are excess alcohol intake, viral hepatitis and non-alcoholic fatty liver disease, with the clinical spectrum ranging in severity from hepatic inflammation to cirrhosis, liver failure or hepatocellular carcinoma (HCC). The genome of HCC exhibits diverse mutational signatures, resulting in recurrent mutations across more than 30 cancer genes1-7. Stem cells from normal livers have a low mutational burden and limited diversity of signatures8, which suggests that the complexity of HCC arises during the progression to chronic liver disease and subsequent malignant transformation. Here, by sequencing whole genomes of 482 microdissections of 100-500 hepatocytes from 5 normal and 9 cirrhotic livers, we show that cirrhotic liver has a higher mutational burden than normal liver. Although rare in normal hepatocytes, structural variants, including chromothripsis, were prominent in cirrhosis. Driver mutations, such as point mutations and structural variants, affected 1-5% of clones. Clonal expansions of millimetres in diameter occurred in cirrhosis, with clones sequestered by the bands of fibrosis that surround regenerative nodules. Some mutational signatures were universal and equally active in both non-malignant hepatocytes and HCCs; some were substantially more active in HCCs than chronic liver disease; and others-arising from exogenous exposures-were present in a subset of patients. The activity of exogenous signatures between adjacent cirrhotic nodules varied by up to tenfold within each patient, as a result of clone-specific and microenvironmental forces. Synchronous HCCs exhibited the same mutational signatures as background cirrhotic liver, but with higher burden. Somatic mutations chronicle the exposures, toxicity, regeneration and clonal structure of liver tissue as it progresses from health to disease.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Totoki Y, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–73. - PubMed
-
- Fujimoto A, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
