

Pan-cancer whole-genome analyses of metastatic solid tumours
- PMID: 31645765
- PMCID: PMC6872491
- DOI: 10.1038/s41586-019-1689-y
Pan-cancer whole-genome analyses of metastatic solid tumours
Abstract
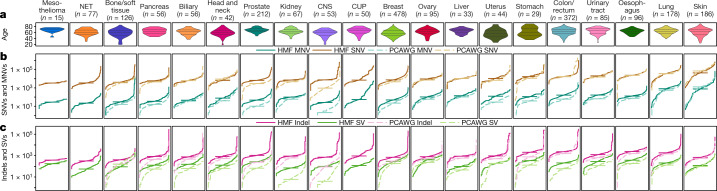
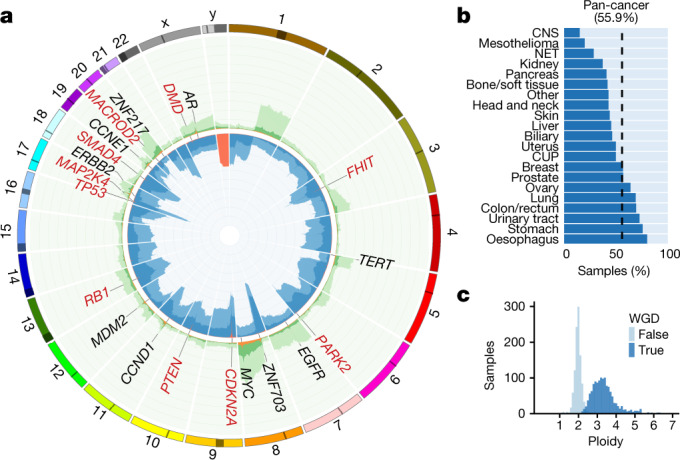
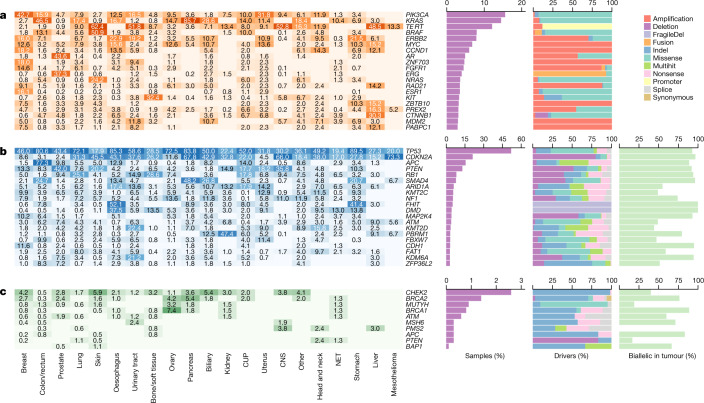
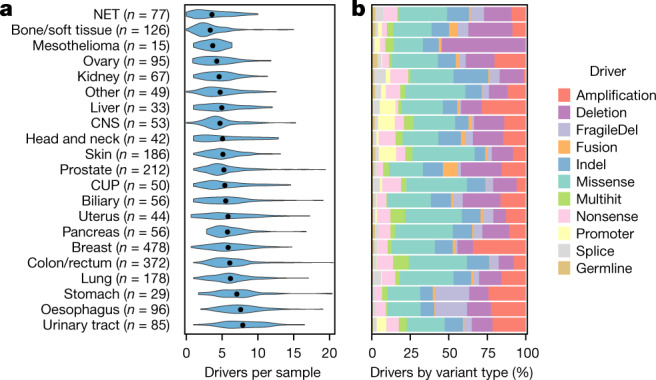
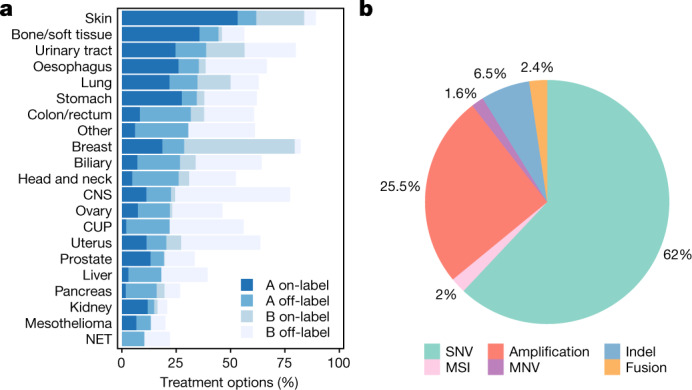
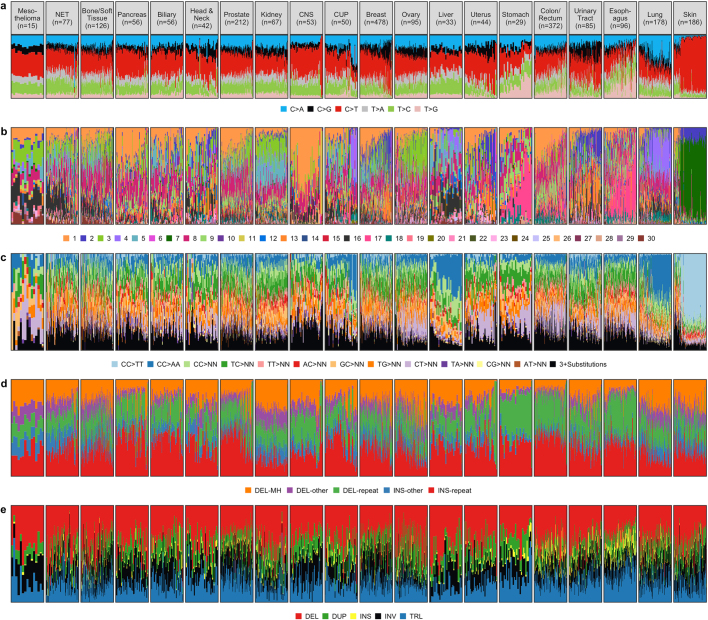
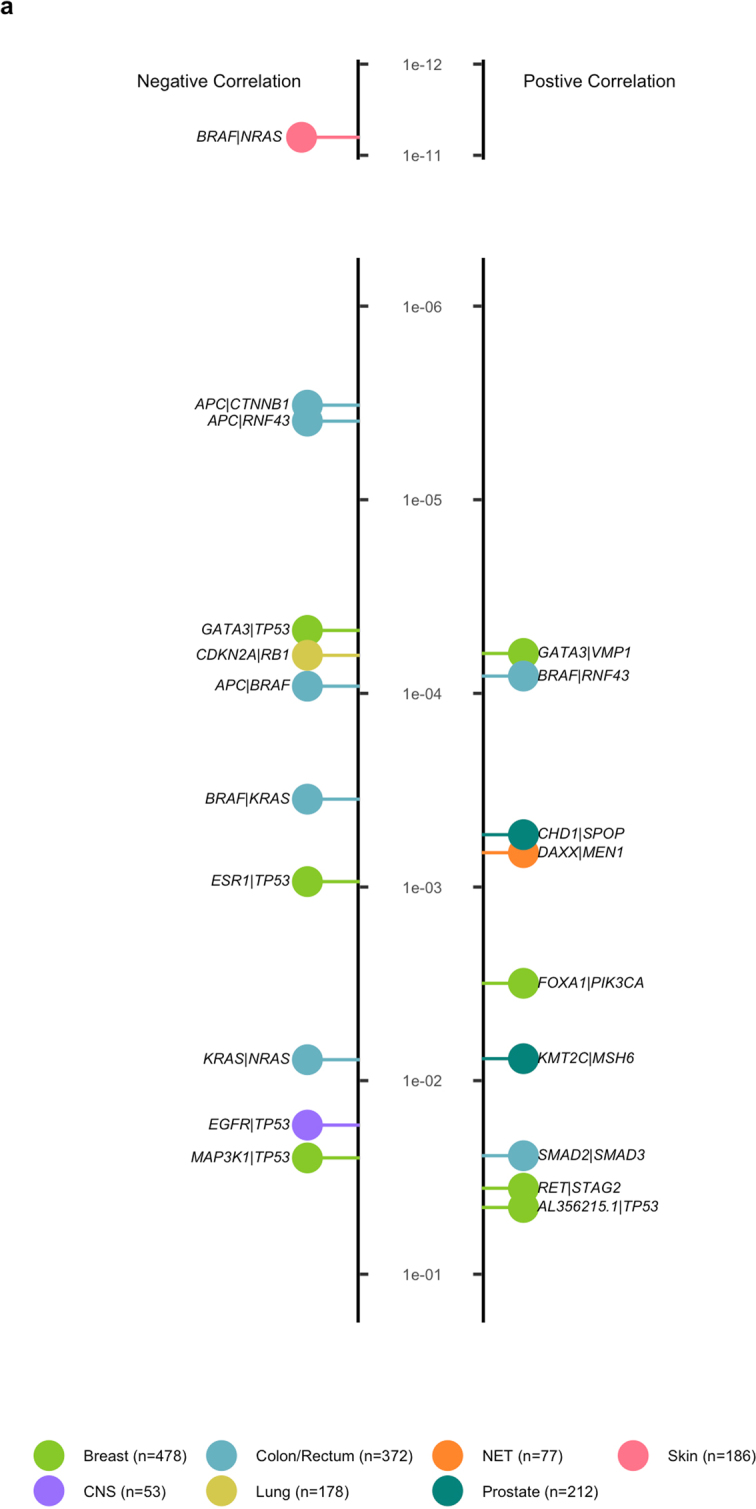
Metastatic cancer is a major cause of death and is associated with poor treatment efficacy. A better understanding of the characteristics of late-stage cancer is required to help adapt personalized treatments, reduce overtreatment and improve outcomes. Here we describe the largest, to our knowledge, pan-cancer study of metastatic solid tumour genomes, including whole-genome sequencing data for 2,520 pairs of tumour and normal tissue, analysed at median depths of 106× and 38×, respectively, and surveying more than 70 million somatic variants. The characteristic mutations of metastatic lesions varied widely, with mutations that reflect those of the primary tumour types, and with high rates of whole-genome duplication events (56%). Individual metastatic lesions were relatively homogeneous, with the vast majority (96%) of driver mutations being clonal and up to 80% of tumour-suppressor genes being inactivated bi-allelically by different mutational mechanisms. Although metastatic tumour genomes showed similar mutational landscape and driver genes to primary tumours, we find characteristics that could contribute to responsiveness to therapy or resistance in individual patients. We implement an approach for the review of clinically relevant associations and their potential for actionability. For 62% of patients, we identify genetic variants that may be used to stratify patients towards therapies that either have been approved or are in clinical trials. This demonstrates the importance of comprehensive genomic tumour profiling for precision medicine in cancer.
Conflict of interest statement
E.E.V. is a supervisory board member of the Hartwig Medical Foundation.
Figures









Comment in
-
Huge whole-genome study of human metastatic cancers.Nature. 2019 Nov;575(7781):60-61. doi: 10.1038/d41586-019-03123-0. Nature. 2019. PMID: 31690848 No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
