

Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer
- PMID: 31649109
- PMCID: PMC6954322
- DOI: 10.1158/2159-8290.CD-19-1006
Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer
Abstract
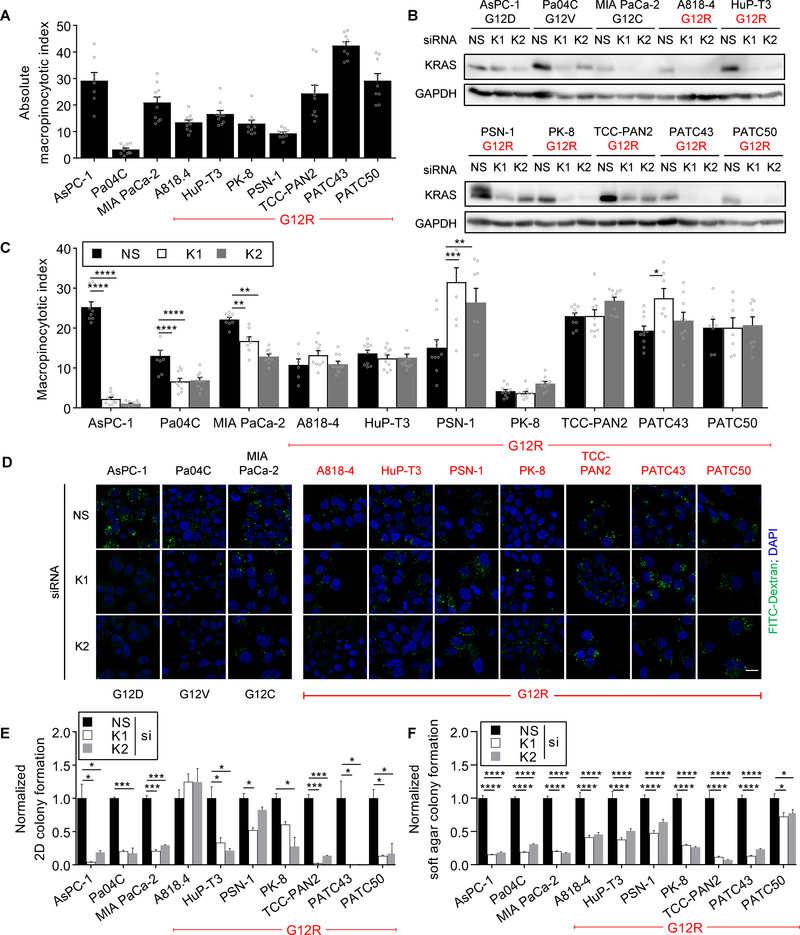
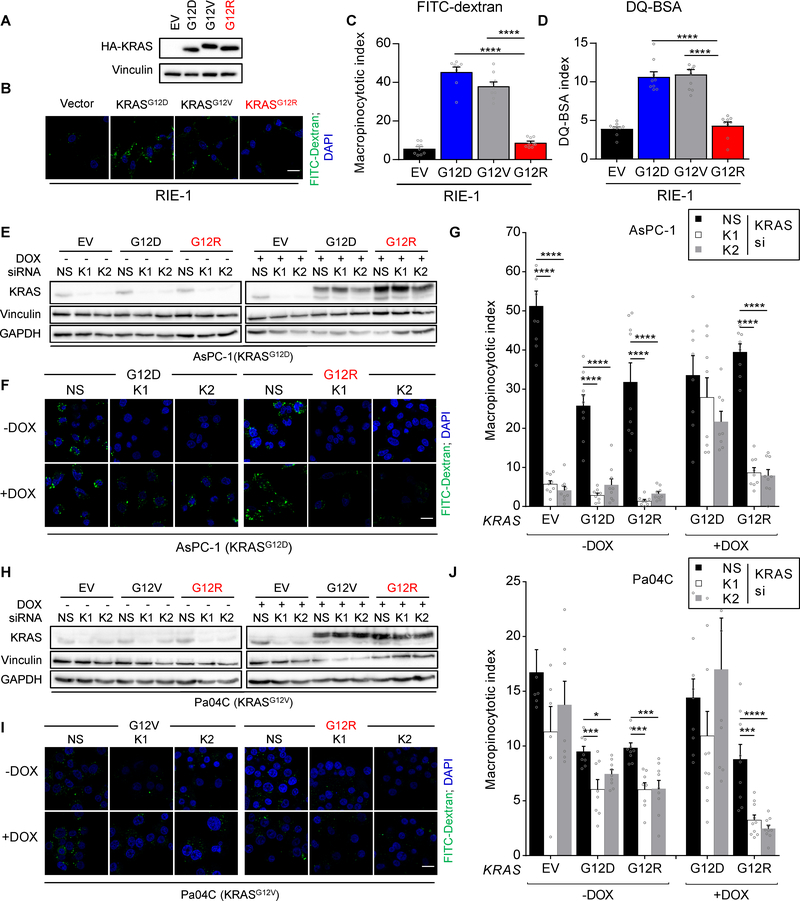
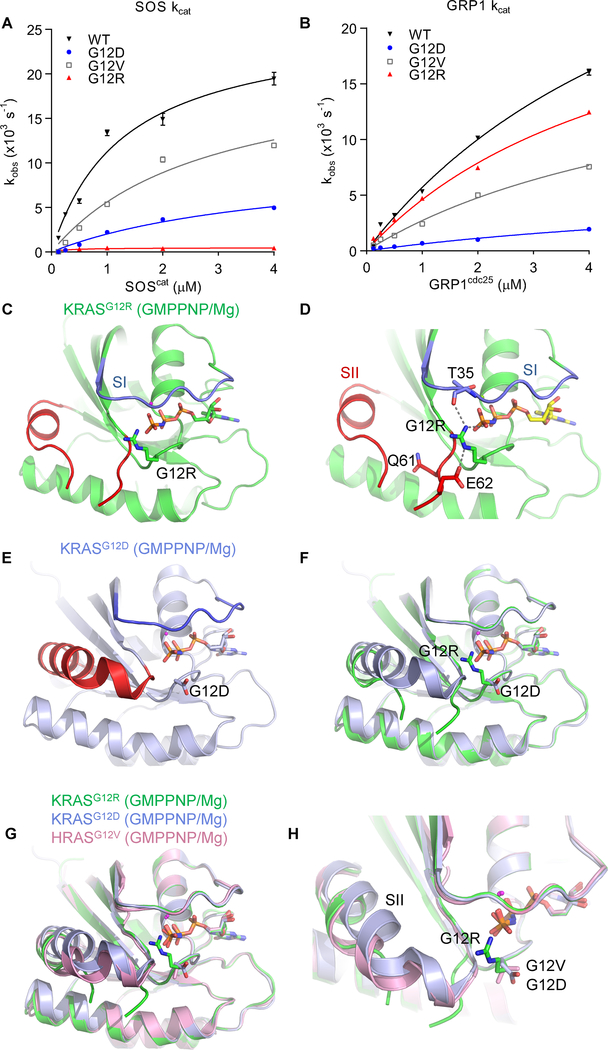
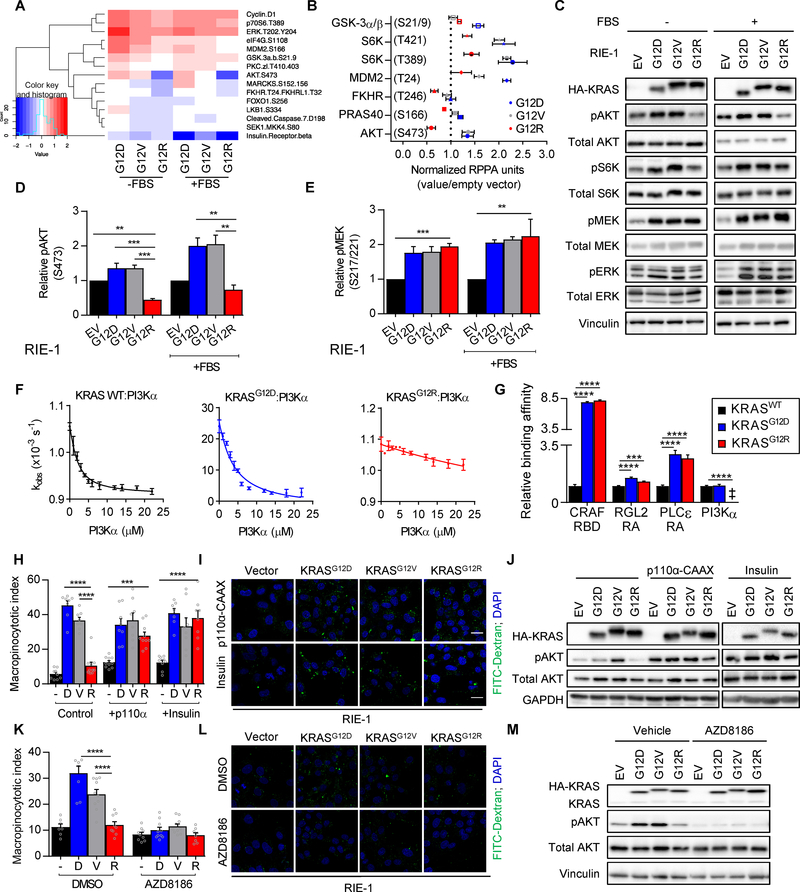
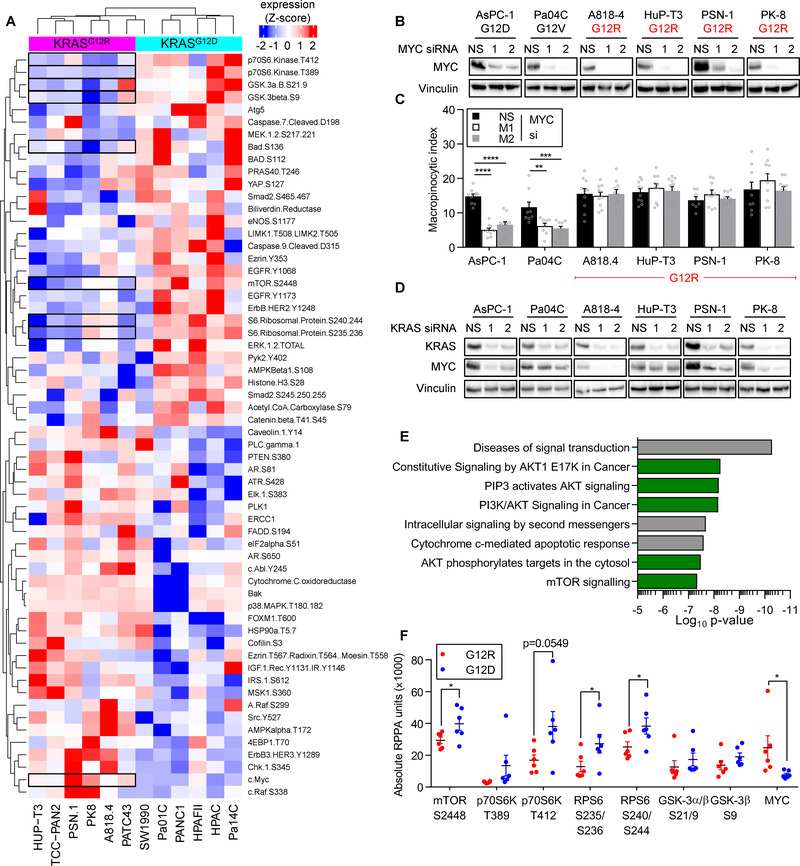
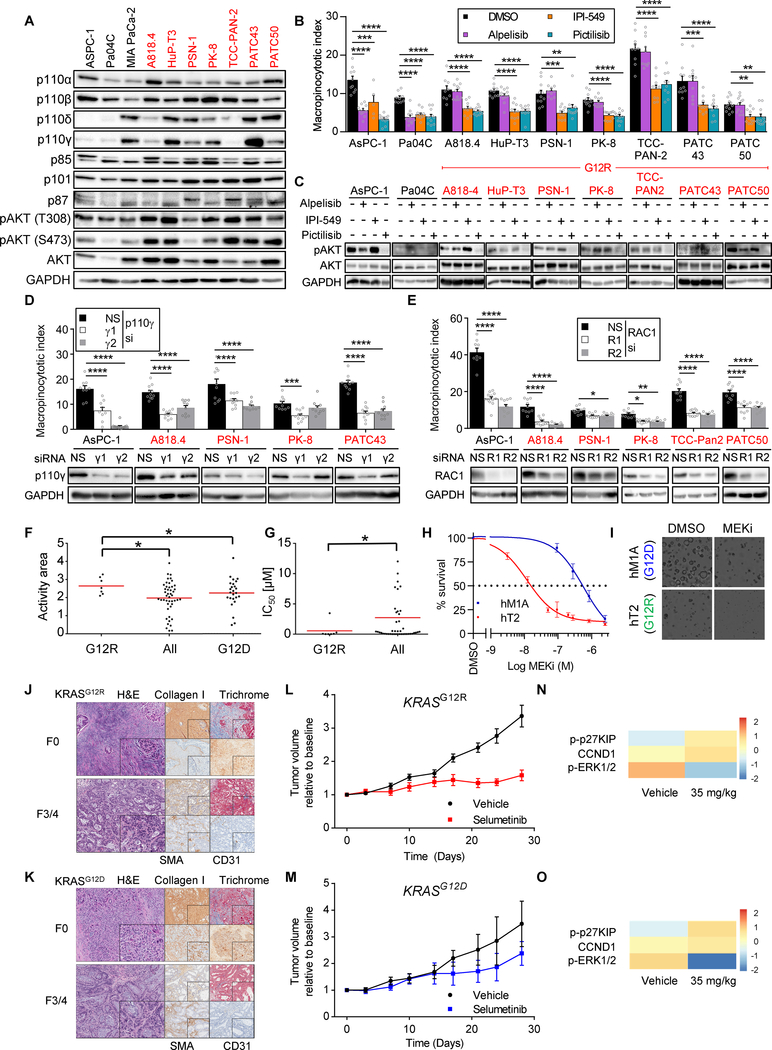
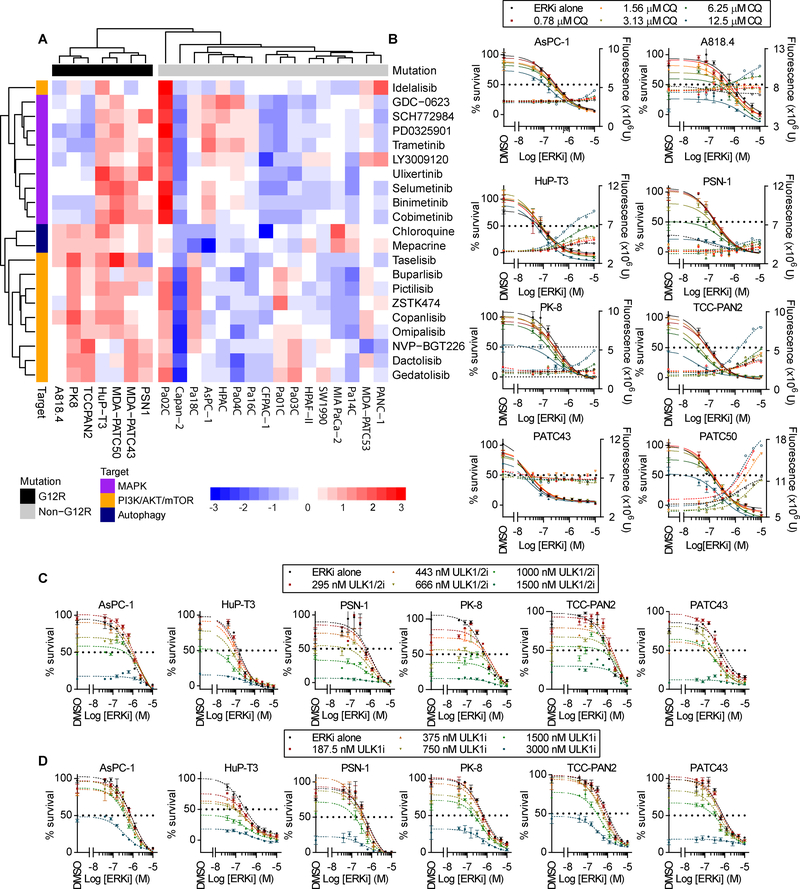
Allele-specific signaling by different KRAS alleles remains poorly understood. The KRAS G12R mutation displays uneven prevalence among cancers that harbor the highest occurrence of KRAS mutations: It is rare (∼1%) in lung and colorectal cancers, yet relatively common (∼20%) in pancreatic ductal adenocarcinoma (PDAC), suggesting context-specific properties. We evaluated whether KRASG12R is functionally distinct from the more common KRASG12D- or KRASG12V-mutant proteins (KRASG12D/V). We found that KRASG12D/V but not KRASG12R drives macropinocytosis and that MYC is essential for macropinocytosis in KRASG12D/V- but not KRASG12R-mutant PDAC. Surprisingly, we found that KRASG12R is defective for interaction with a key effector, p110α PI3K (PI3Kα), due to structural perturbations in switch II. Instead, upregulated KRAS-independent PI3Kγ activity was able to support macropinocytosis in KRASG12R-mutant PDAC. Finally, we determined that KRASG12R-mutant PDAC displayed a distinct drug sensitivity profile compared with KRASG12D-mutant PDAC but is still responsive to the combined inhibition of ERK and autophagy. SIGNIFICANCE: We determined that KRASG12R is impaired in activating a key effector, p110α PI3K. As such, KRASG12R is impaired in driving macropinocytosis. However, overexpression of PI3Kγ in PDAC compensates for this deficiency, providing one basis for the prevalence of this otherwise rare KRAS mutant in pancreatic cancer but not other cancers.See related commentary by Falcomatà et al., p. 23.This article is highlighted in the In This Issue feature, p. 1.
©2019 American Association for Cancer Research.
Figures
Comment in
-
Personalizing KRAS-Mutant Allele-Specific Therapies.Cancer Discov. 2020 Jan;10(1):23-25. doi: 10.1158/2159-8290.CD-19-1261. Cancer Discov. 2020. PMID: 31919121
-
The type of KRAS mutation drives PI3Kα/γ signalling dependency: Implication for the choice of targeted therapy in pancreatic adenocarcinoma patients.Clin Res Hepatol Gastroenterol. 2021 Jan;45(1):101473. doi: 10.1016/j.clinre.2020.05.021. Epub 2020 Jun 24. Clin Res Hepatol Gastroenterol. 2021. PMID: 32593694
-
Distinct metabolic preference of atypical KRAS mutant.Ann Transl Med. 2020 Oct;8(20):1326. doi: 10.21037/atm-20-3252. Ann Transl Med. 2020. PMID: 33209906 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- U01 CA224146/CA/NCI NIH HHS/United States
- R01 CA042978/CA/NCI NIH HHS/United States
- T32 CA009156/CA/NCI NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- K08 CA218420/CA/NCI NIH HHS/United States
- U01 CA199235/CA/NCI NIH HHS/United States
- T32 CA071341/CA/NCI NIH HHS/United States
- F31 CA180628/CA/NCI NIH HHS/United States
- F32 CA200313/CA/NCI NIH HHS/United States
- P50 CA127003/CA/NCI NIH HHS/United States
- P30 AI050410/AI/NIAID NIH HHS/United States
- P41 GM103403/GM/NIGMS NIH HHS/United States
- F32 CA221005/CA/NCI NIH HHS/United States
- P01 CA203657/CA/NCI NIH HHS/United States
- R35 CA232113/CA/NCI NIH HHS/United States
- P50 CA196510/CA/NCI NIH HHS/United States
- F30 CA243253/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
