

Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia
- PMID: 31652573
- PMCID: PMC6834160
- DOI: 10.3390/ijms20205236
Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia
Abstract
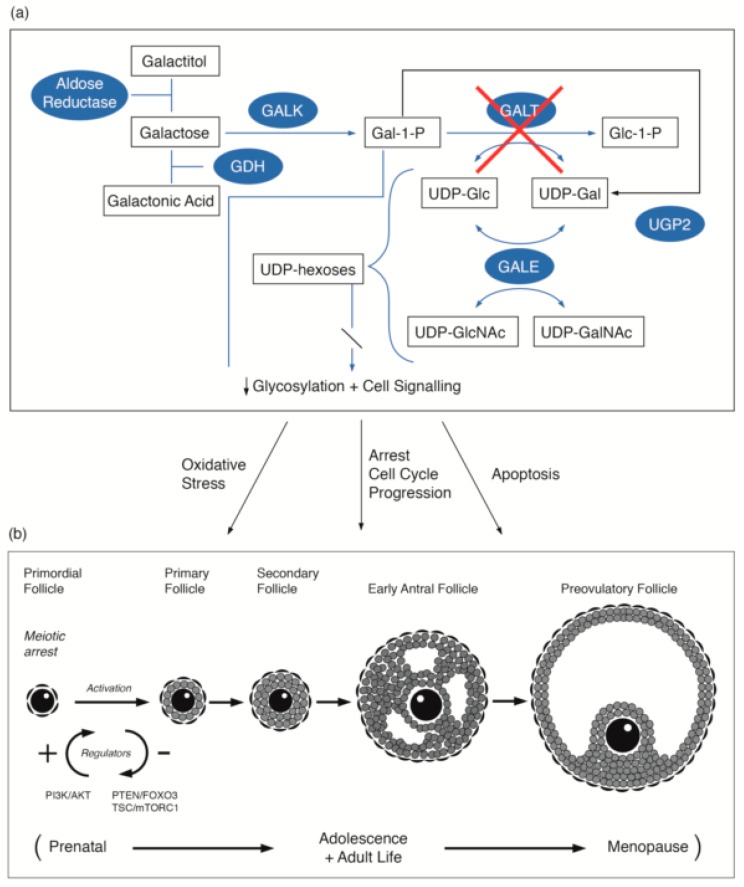
Classical galactosaemia (CG) (OMIM 230400) is a rare inborn error of galactose metabolism caused by the deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT, EC 2.7.7.12). Primary ovarian insufficiency (POI) is the most common long-term complication experienced by females with CG, presenting with hypergonadotrophic hypoestrogenic infertility affecting at least 80% of females despite new-born screening and lifelong galactose dietary restriction. In this review, we describe the hypothesized pathophysiology of POI from CG, implications of timing of the ovarian dysfunction, and the new horizons and future prospects for treatments and fertility preservation.
Keywords: classical galactosaemia; fertility preservation; pathophysiology; primary ovarian insufficiency.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Valle D., Antonarakis S., Ballabio A., Beaudet A., Mitchell G.A. The Online Metabolic and Molecular Bases of Inherited Disease. The McGraw-Hill Companies; New York, NY, USA: 2008.
-
- Berry G.T. Classic Galactosemia and Clinical Variant Galactosemia. [(accessed on 10 September 2019)];Gene Rev. 2017 Available online: https://www.ncbi.nlm.nih.gov/books/NBK1518/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
