

Chaperone-Mediated Autophagy in the Liver: Good or Bad?
- PMID: 31652893
- PMCID: PMC6912708
- DOI: 10.3390/cells8111308
Chaperone-Mediated Autophagy in the Liver: Good or Bad?
Abstract
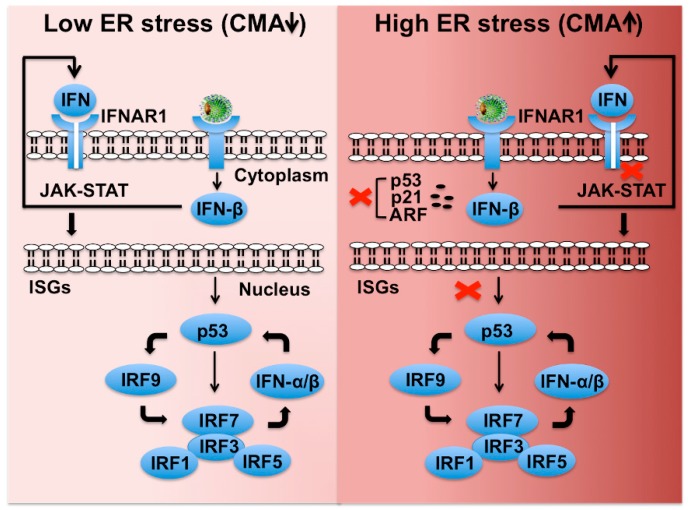
Hepatitis C virus (HCV) infection triggers autophagy processes, which help clear out the dysfunctional viral and cellular components that would otherwise inhibit the virus replication. Increased cellular autophagy may kill the infected cell and terminate the infection without proper regulation. The mechanism of autophagy regulation during liver disease progression in HCV infection is unclear. The autophagy research has gained a lot of attention recently since autophagy impairment is associated with the development of hepatocellular carcinoma (HCC). Macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) are three autophagy processes involved in the lysosomal degradation and extracellular release of cytosolic cargoes under excessive stress. Autophagy processes compensate for each other during extreme endoplasmic reticulum (ER) stress to promote host and microbe survival as well as HCC development in the highly stressed microenvironment of the cirrhotic liver. This review describes the molecular details of how excessive cellular stress generated during HCV infection activates CMA to improve cell survival. The pathological implications of stress-related CMA activation resulting in the loss of hepatic innate immunity and tumor suppressors, which are most often observed among cirrhotic patients with HCC, are discussed. The oncogenic cell programming through autophagy regulation initiated by a cytoplasmic virus may facilitate our understanding of HCC mechanisms related to non-viral etiologies and metabolic conditions such as uncontrolled type II diabetes. We propose that a better understanding of how excessive cellular stress leads to cancer through autophagy modulation may allow therapeutic development and early detection of HCC.
Keywords: chaperone-mediated autophagy (CMA); endoplasmic reticulum stress (ER stress); hepatitis C virus (HCV); hepatocellular carcinoma (HCC); interferon alpha receptor 1 (IFNAR1); nuclear factor erythroid 2-related factor 2 (NRF2).
Conflict of interest statement
The authors declare no conflict of interest.
Figures







Similar articles
-
Integrated stress response in hepatitis C promotes Nrf2-related chaperone-mediated autophagy: A novel mechanism for host-microbe survival and HCC development in liver cirrhosis.Semin Cell Dev Biol. 2020 May;101:20-35. doi: 10.1016/j.semcdb.2019.07.015. Epub 2019 Aug 8. Semin Cell Dev Biol. 2020. PMID: 31386899 Free PMC article. Review.
-
Hepatitis C Virus NS5A Protein Promotes the Lysosomal Degradation of Hepatocyte Nuclear Factor 1α via Chaperone-Mediated Autophagy.J Virol. 2018 Jun 13;92(13):e00639-18. doi: 10.1128/JVI.00639-18. Print 2018 Jul 1. J Virol. 2018. PMID: 29695419 Free PMC article.
-
Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response.Viruses. 2016 May 23;8(5):150. doi: 10.3390/v8050150. Viruses. 2016. PMID: 27223299 Free PMC article. Review.
-
Chaperone-Mediated Autophagy Promotes Beclin1 Degradation in Persistently Infected Hepatitis C Virus Cell Culture.Am J Pathol. 2018 Oct;188(10):2339-2355. doi: 10.1016/j.ajpath.2018.06.022. Epub 2018 Aug 1. Am J Pathol. 2018. PMID: 30075149 Free PMC article.
-
Chaperone-Mediated Autophagy Targets IFNAR1 for Lysosomal Degradation in Free Fatty Acid Treated HCV Cell Culture.PLoS One. 2015 May 11;10(5):e0125962. doi: 10.1371/journal.pone.0125962. eCollection 2015. PLoS One. 2015. PMID: 25961570 Free PMC article.
Cited by
-
The Role of Chaperone-Mediated Autophagy in Hepatitis C Virus-Induced Pathogenesis.Front Cell Infect Microbiol. 2021 Dec 2;11:796664. doi: 10.3389/fcimb.2021.796664. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 34926330 Free PMC article. Review.
-
The Role of Macroautophagy and Chaperone-Mediated Autophagy in the Pathogenesis and Management of Hepatocellular Carcinoma.Cancers (Basel). 2022 Feb 1;14(3):760. doi: 10.3390/cancers14030760. Cancers (Basel). 2022. PMID: 35159028 Free PMC article. Review.
-
EnsembleDL-ATG: Identifying autophagy proteins by integrating their sequence and evolutionary information using an ensemble deep learning framework.Comput Struct Biotechnol J. 2023 Sep 29;21:4836-4848. doi: 10.1016/j.csbj.2023.09.036. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 37854634 Free PMC article.
-
Developing and Validating an Autophagy Gene-Set-Based Prognostic Signature in Hepatocellular Carcinoma Patients.Int J Gen Med. 2022 Nov 28;15:8399-8415. doi: 10.2147/IJGM.S388592. eCollection 2022. Int J Gen Med. 2022. PMID: 36465273 Free PMC article.
-
Higher pNRF2, SOCS3, IRF3, and RIG1 Tissue Protein Expression in NASH Patients versus NAFL Patients: pNRF2 Expression Is Concomitantly Associated with Elevated Fasting Glucose Levels.J Pers Med. 2023 Jul 18;13(7):1152. doi: 10.3390/jpm13071152. J Pers Med. 2023. PMID: 37511764 Free PMC article.
References
-
- Stanaway J.D., Flaxman A.D., Naghavi M., Fitzmaurice C., Vos T., Abubakar I., Abu-Raddad L.J., Assadi R., Bhala N., Cowie B., et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet. 2016;388:1081–1088. doi: 10.1016/S0140-6736(16)30579-7. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
