

GenGraph: a python module for the simple generation and manipulation of genome graphs
- PMID: 31653197
- PMCID: PMC6894214
- DOI: 10.1186/s12859-019-3115-8
GenGraph: a python module for the simple generation and manipulation of genome graphs
Abstract
Background: As sequencing technology improves, the concept of a single reference genome is becoming increasingly restricting. In the case of Mycobacterium tuberculosis, one must often choose between using a genome that is closely related to the isolate, or one that is annotated in detail. One promising solution to this problem is through the graph based representation of collections of genomes as a single genome graph. Though there are currently a handful of tools that can create genome graphs and have demonstrated the advantages of this new paradigm, there still exists a need for flexible tools that can be used by researchers to overcome challenges in genomics studies.
Results: We present GenGraph, a Python toolkit and accompanying modules that use existing multiple sequence alignment tools to create genome graphs. Python is one of the most popular coding languages for the biological sciences, and by providing these tools, GenGraph makes it easier to experiment and develop new tools that utilise genome graphs. The conceptual model used is highly intuitive, and as much as possible the graph structure represents the biological relationship between the genomes. This design means that users will quickly be able to start creating genome graphs and using them in their own projects. We outline the methods used in the generation of the graphs, and give some examples of how the created graphs may be used. GenGraph utilises existing file formats and methods in the generation of these graphs, allowing graphs to be visualised and imported with widely used applications, including Cytoscape, R, and Java Script.
Conclusions: GenGraph provides a set of tools for generating graph based representations of sets of sequences with a simple conceptual model, written in the widely used coding language Python, and publicly available on Github.
Keywords: Genome; Graph; Module; Python; Toolkit.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
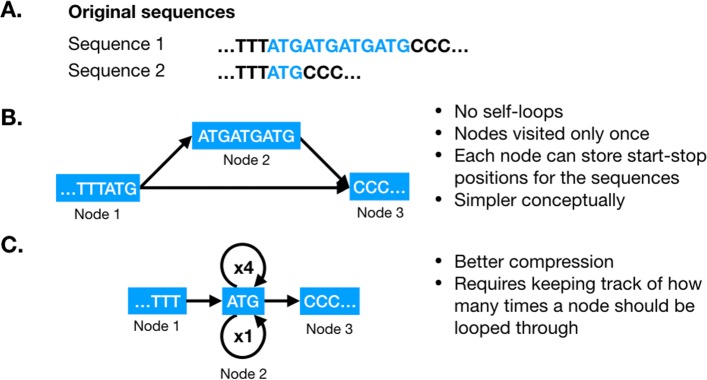

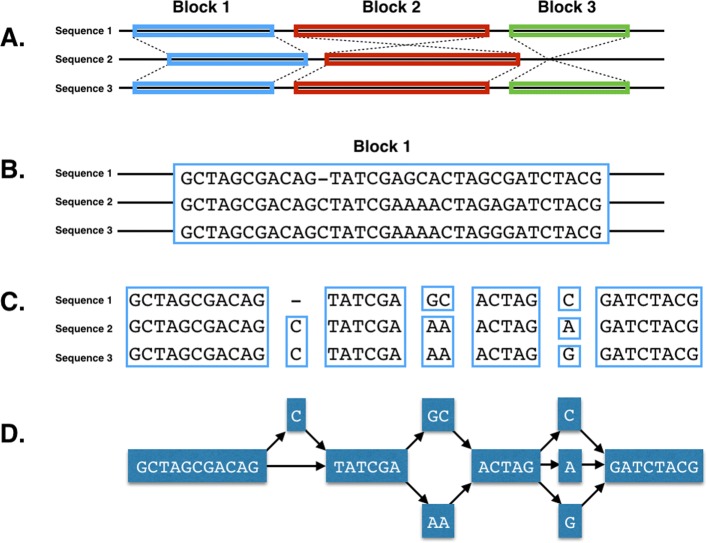
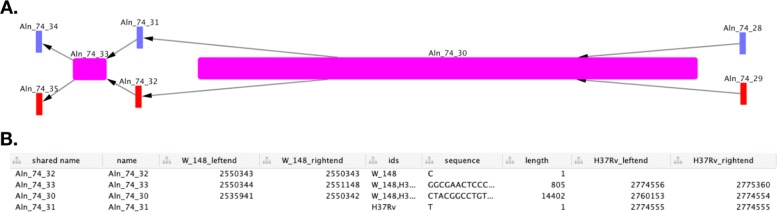
References
-
- VG Team. Variant Graph. https://github.com/vgteam/vg/. Accessed 10 Dec 2018.
-
- Sheikhizadeh S, Schranz ME, Akdel M, de Ridder D, Smit S. PanTools: representation, storage and exploration of pan-genomic data. Bioinformatics. 2016; 32(17):487–93. 10.1093/bioinformatics/btw455. - PubMed
-
- Gonnella G, Kurtz S. GfaPy: A flexible and extensible software library for handling sequence graphs in Python. Bioinformatics. 2017; 33(19):3094–5. 10.1093/bioinformatics/btx398. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
