

The Biology of B-Progenitor Acute Lymphoblastic Leukemia
- PMID: 31653664
- PMCID: PMC7328455
- DOI: 10.1101/cshperspect.a034835
The Biology of B-Progenitor Acute Lymphoblastic Leukemia
Abstract
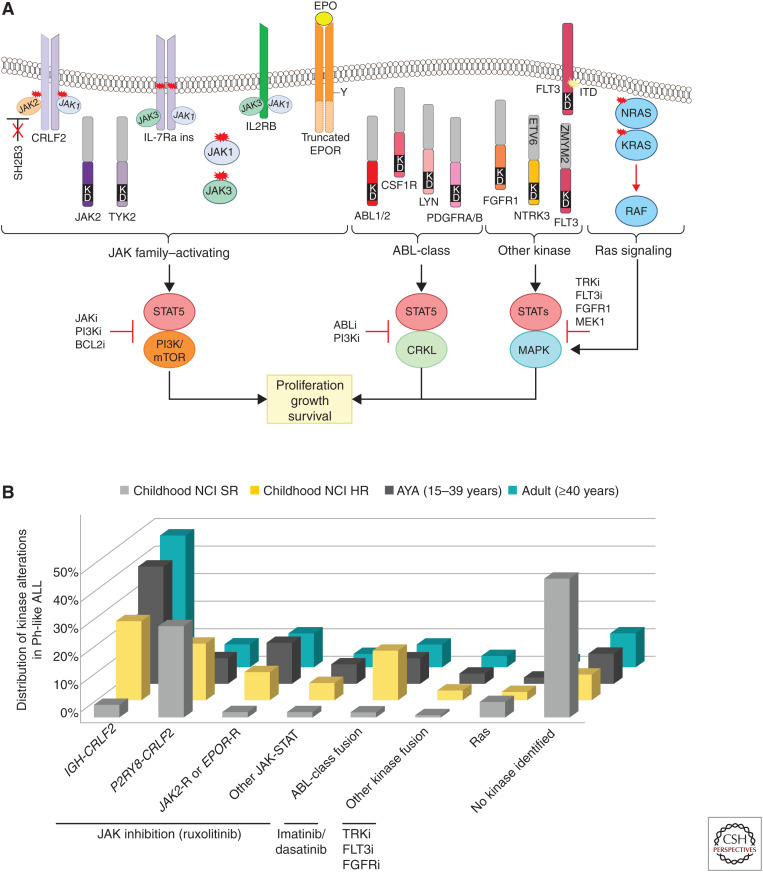
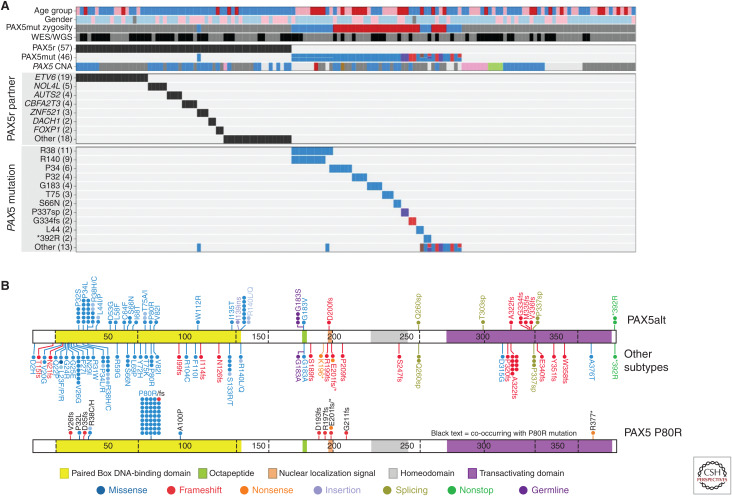
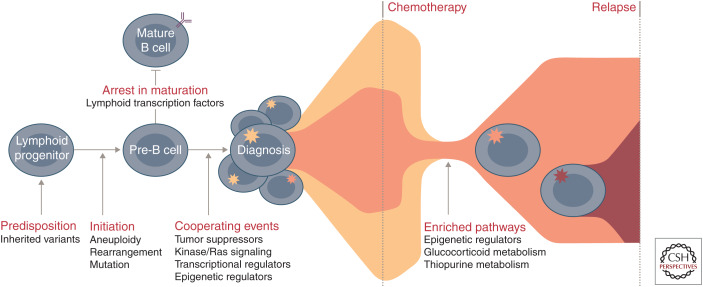
Genomic analyses have revolutionized our understanding of the biology of B-progenitor acute lymphoblastic leukemia (ALL). Studies of thousands of cases across the age spectrum have revised the taxonomy of B-ALL by identifying multiple new subgroups with diverse sequence and structural initiating events that vary substantially by age at diagnosis and prognostic significance. There is a growing appreciation of the role of inherited genetic variation in predisposition to ALL and drug responsiveness and of the nature of genetic variegation and clonal evolution that may be targeted for improved diagnostic, risk stratification, disease monitoring, and therapeutic intervention. This review provides an overview of the current state of knowledge of the genetic basis of B-ALL, with an emphasis on recent discoveries that have changed our approach to diagnosis and monitoring.
Copyright © 2020 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures

References
-
- Agraz Doblas A, Bueno C, Bashford-Rogers R, Roy A, Schneider P, Bardini M, Ballerini P, Cazzaniga G, Moreno T, Revilla C, et al. 2019. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 104: 1176–1188. 10.3324/haematol.2018.206375 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical