

Scalable Data Analysis in Proteomics and Metabolomics Using BioContainers and Workflows Engines
- PMID: 31657527
- PMCID: PMC7613303
- DOI: 10.1002/pmic.201900147
Scalable Data Analysis in Proteomics and Metabolomics Using BioContainers and Workflows Engines
Abstract
The recent improvements in mass spectrometry instruments and new analytical methods are increasing the intersection between proteomics and big data science. In addition, bioinformatics analysis is becoming increasingly complex and convoluted, involving multiple algorithms and tools. A wide variety of methods and software tools have been developed for computational proteomics and metabolomics during recent years, and this trend is likely to continue. However, most of the computational proteomics and metabolomics tools are designed as single-tiered software application where the analytics tasks cannot be distributed, limiting the scalability and reproducibility of the data analysis. In this paper the key steps of metabolomics and proteomics data processing, including the main tools and software used to perform the data analysis, are summarized. The combination of software containers with workflows environments for large-scale metabolomics and proteomics analysis is discussed. Finally, a new approach for reproducible and large-scale data analysis based on BioContainers and two of the most popular workflow environments, Galaxy and Nextflow, is introduced to the proteomics and metabolomics communities.
Keywords: bioconda; biocontainers; bioinformatics; containers; large scale data analysis; workflows.
© 2019 The Authors. Proteomics published by WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
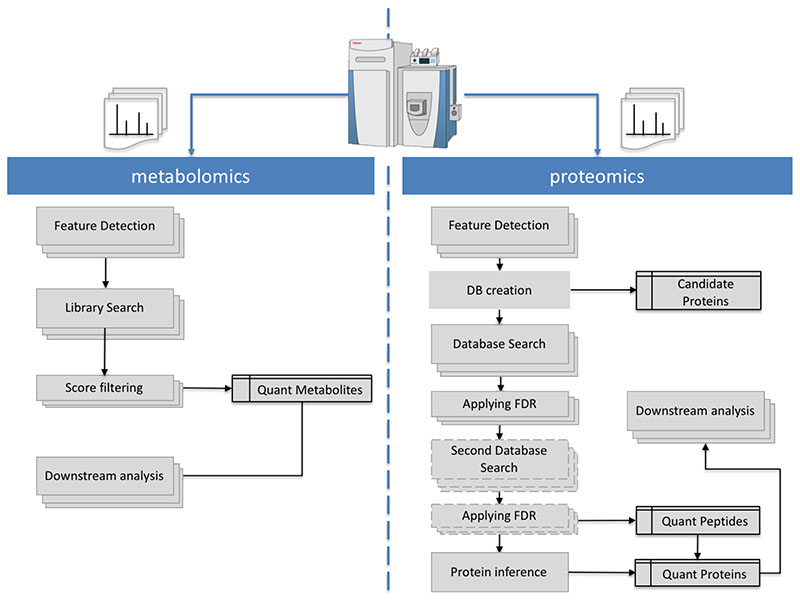
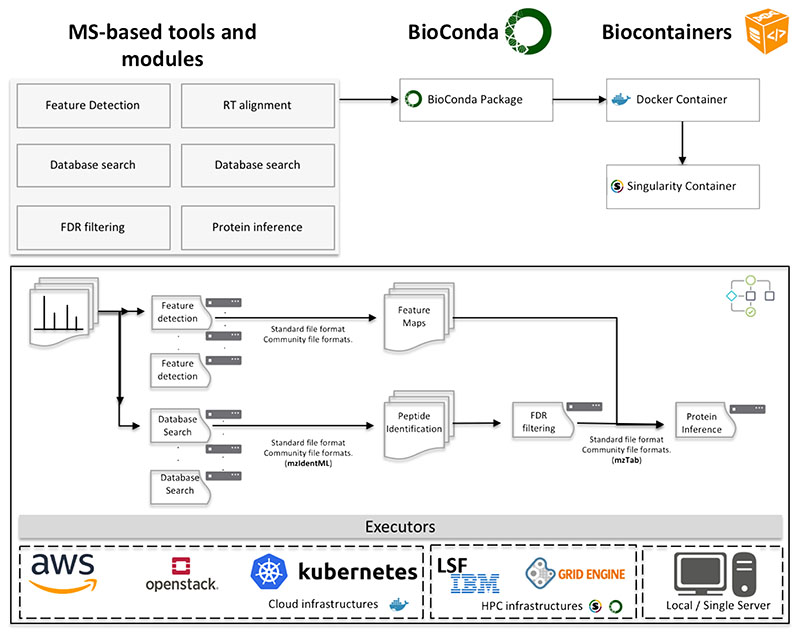
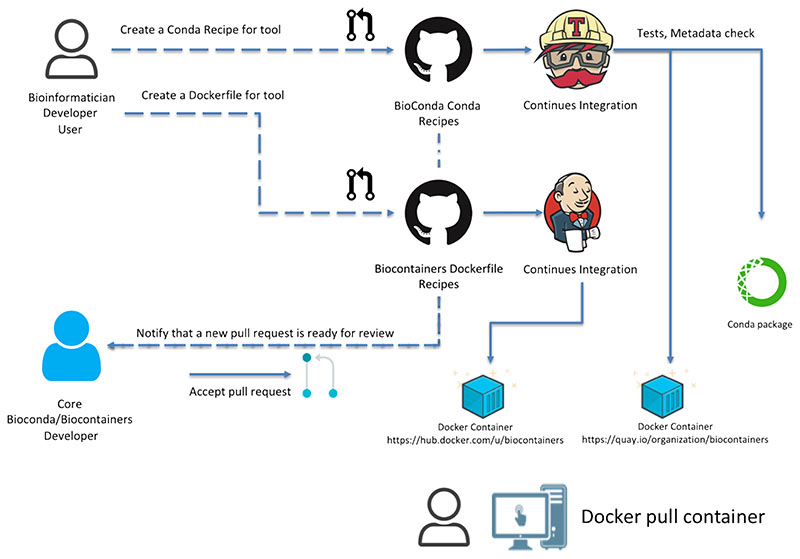
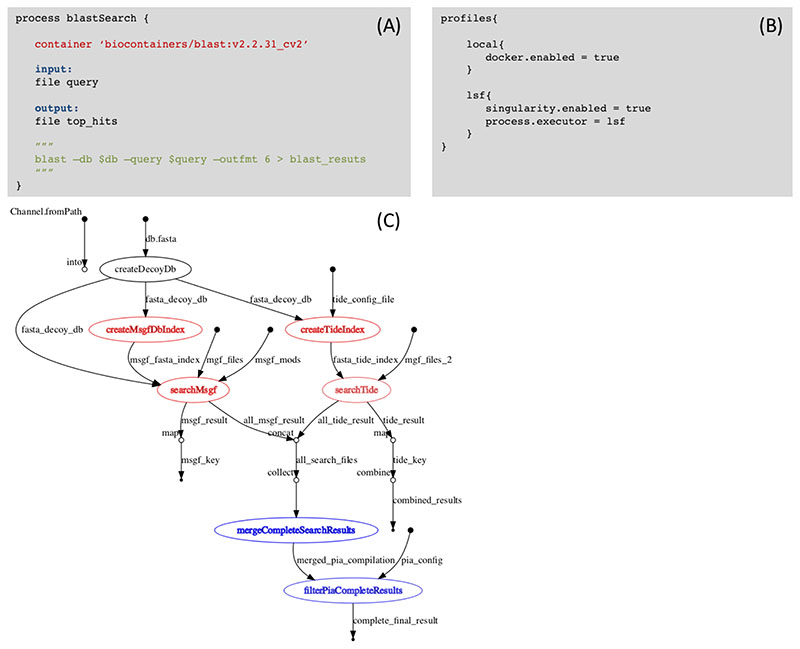

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
