

The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients
- PMID: 31658955
- PMCID: PMC6954325
- DOI: 10.1158/2159-8290.CD-19-1167
The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients
Abstract
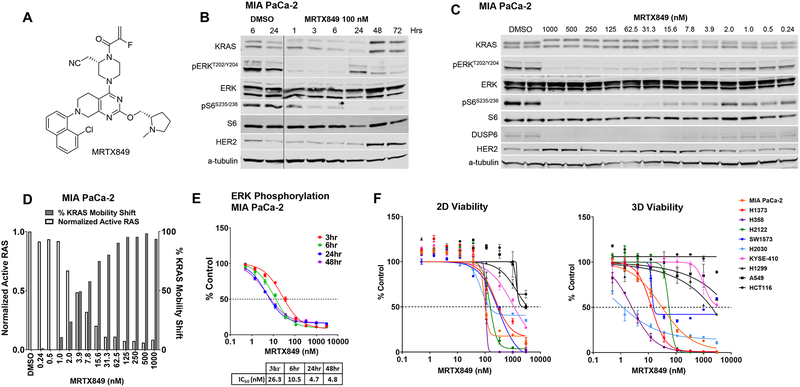
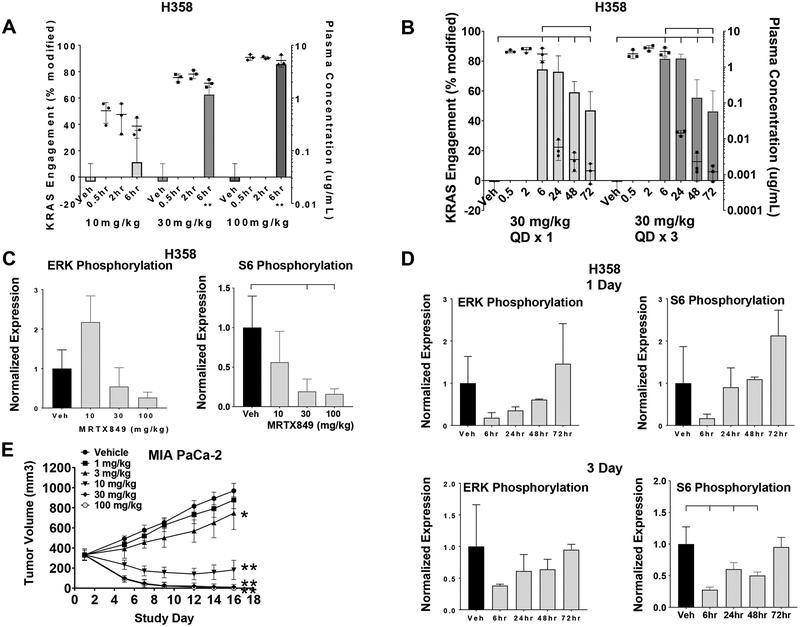
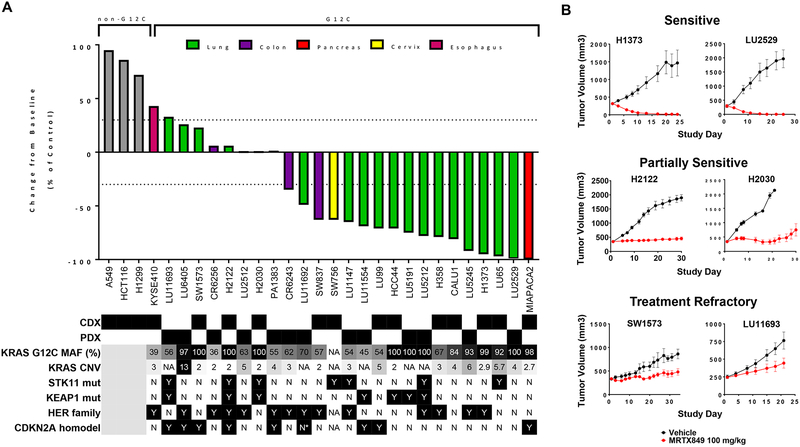
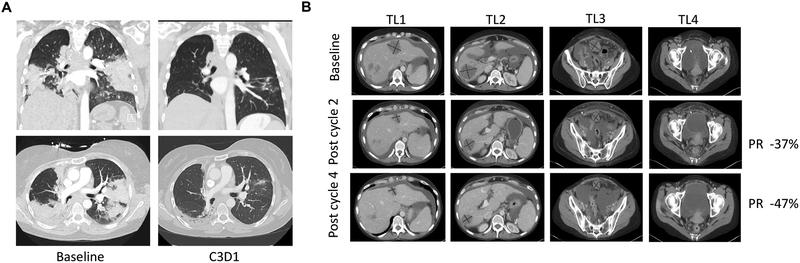
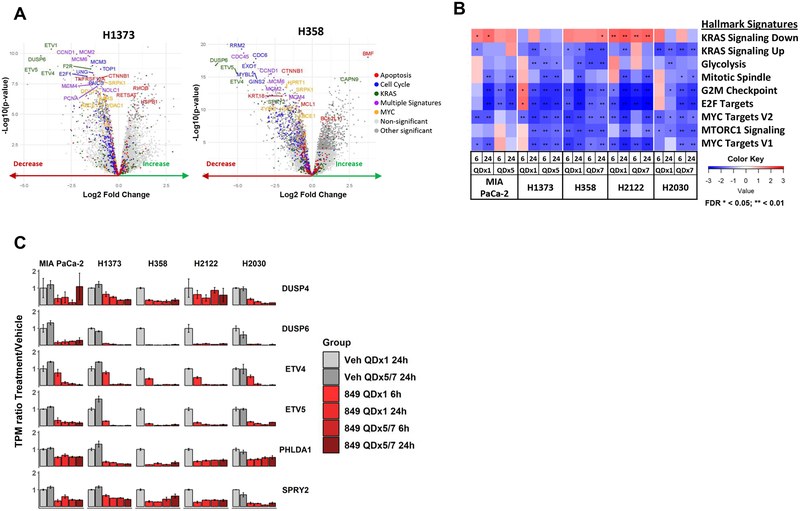
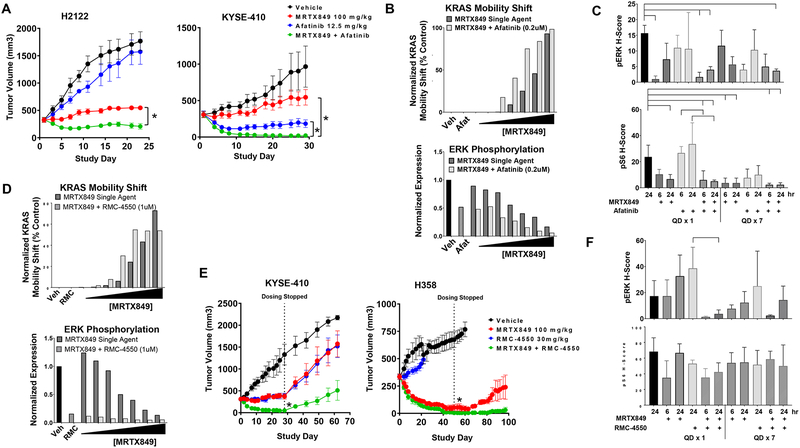
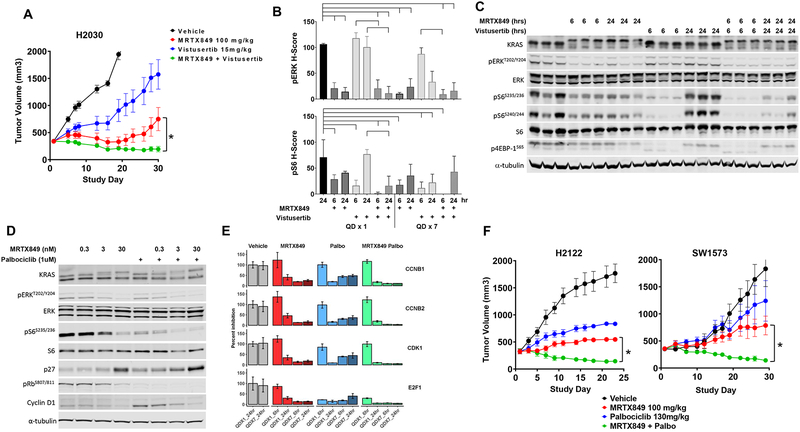
Despite decades of research, efforts to directly target KRAS have been challenging. MRTX849 was identified as a potent, selective, and covalent KRASG12C inhibitor that exhibits favorable drug-like properties, selectively modifies mutant cysteine 12 in GDP-bound KRASG12C, and inhibits KRAS-dependent signaling. MRTX849 demonstrated pronounced tumor regression in 17 of 26 (65%) KRASG12C-positive cell line- and patient-derived xenograft models from multiple tumor types, and objective responses have been observed in patients with KRASG12C-positive lung and colon adenocarcinomas. Comprehensive pharmacodynamic and pharmacogenomic profiling in sensitive and partially resistant nonclinical models identified mechanisms implicated in limiting antitumor activity including KRAS nucleotide cycling and pathways that induce feedback reactivation and/or bypass KRAS dependence. These factors included activation of receptor tyrosine kinases (RTK), bypass of KRAS dependence, and genetic dysregulation of cell cycle. Combinations of MRTX849 with agents that target RTKs, mTOR, or cell cycle demonstrated enhanced response and marked tumor regression in several tumor models, including MRTX849-refractory models. SIGNIFICANCE: The discovery of MRTX849 provides a long-awaited opportunity to selectively target KRASG12C in patients. The in-depth characterization of MRTX849 activity, elucidation of response and resistance mechanisms, and identification of effective combinations provide new insight toward KRAS dependence and the rational development of this class of agents.See related commentary by Klempner and Hata, p. 20.This article is highlighted in the In This Issue feature, p. 1.
©2019 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment in
-
KRAS-G12C in the crosshairs.Nat Rev Cancer. 2020 Jan;20(1):3. doi: 10.1038/s41568-019-0228-3. Nat Rev Cancer. 2020. PMID: 31728026 No abstract available.
-
Two new agents target KRAS G12C.Nat Rev Clin Oncol. 2020 Jan;17(1):6. doi: 10.1038/s41571-019-0304-3. Nat Rev Clin Oncol. 2020. PMID: 31772333 No abstract available.
-
Can the Help Match the Hype? KRASG12C-Specific Inhibitors and Beyond.Cancer Discov. 2020 Jan;10(1):20-22. doi: 10.1158/2159-8290.CD-19-1255. Cancer Discov. 2020. PMID: 31919120
References
-
- John J, Sohmen R, Feuerstein J, Linke R, Wittinghofer A, and Goody RS, Kinetics of interaction of nucleotides with nucleotide-free H-ras p21. Biochemistry, 1990. 29(25): p. 6058–65. - PubMed
-
- Matikas A, Mistriotis D, Georgoulias V, and Kotsakis A, Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit Rev Oncol Hematol, 2017. 110: p. 1–12. - PubMed
-
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al., Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell, 2018. 172(3): p. 578–589 e17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
