

A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers
- PMID: 31659164
- PMCID: PMC6817842
- DOI: 10.1038/s41467-019-12812-3
A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers
Abstract
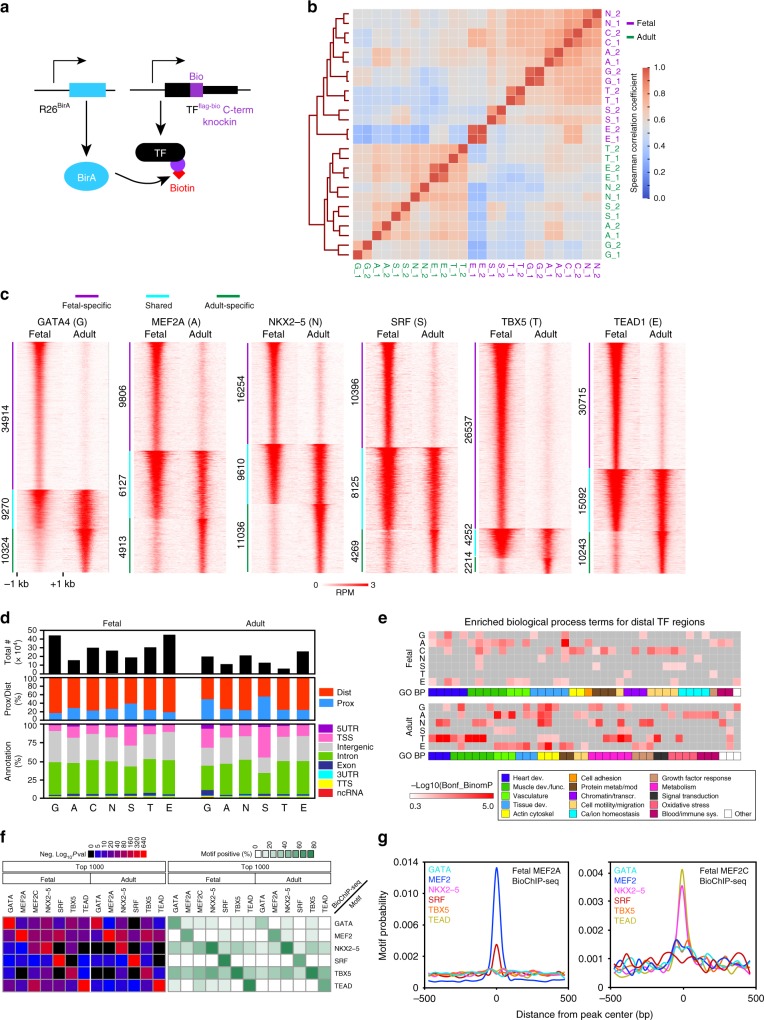
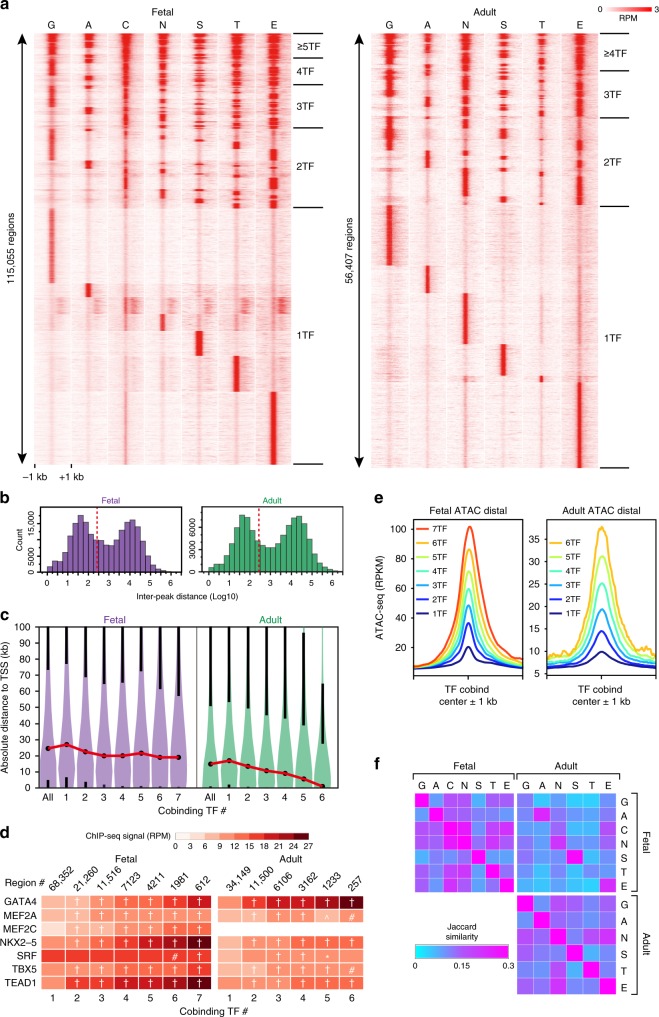
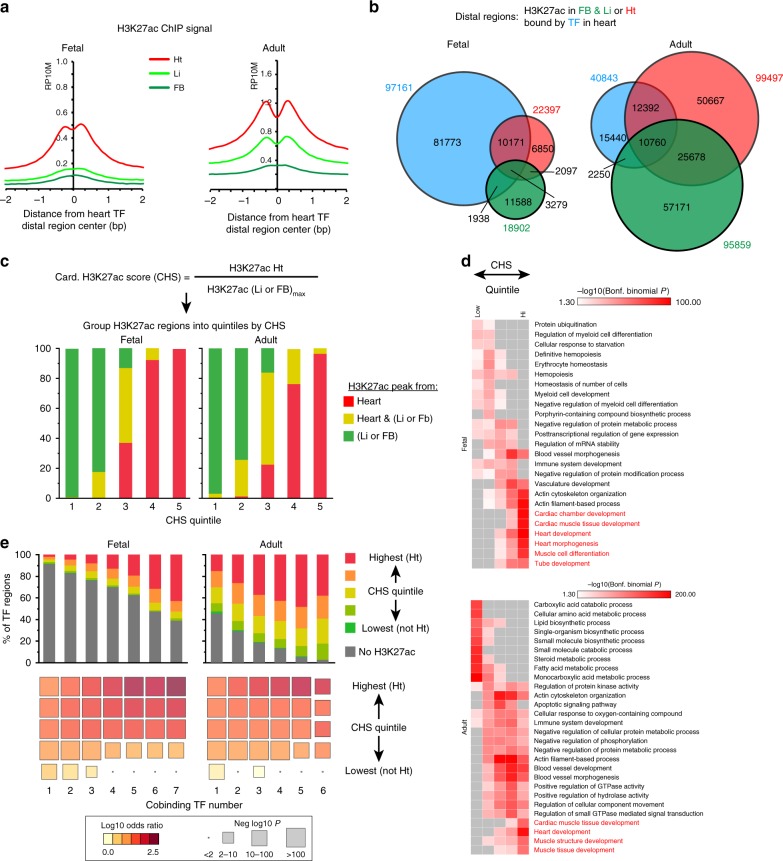
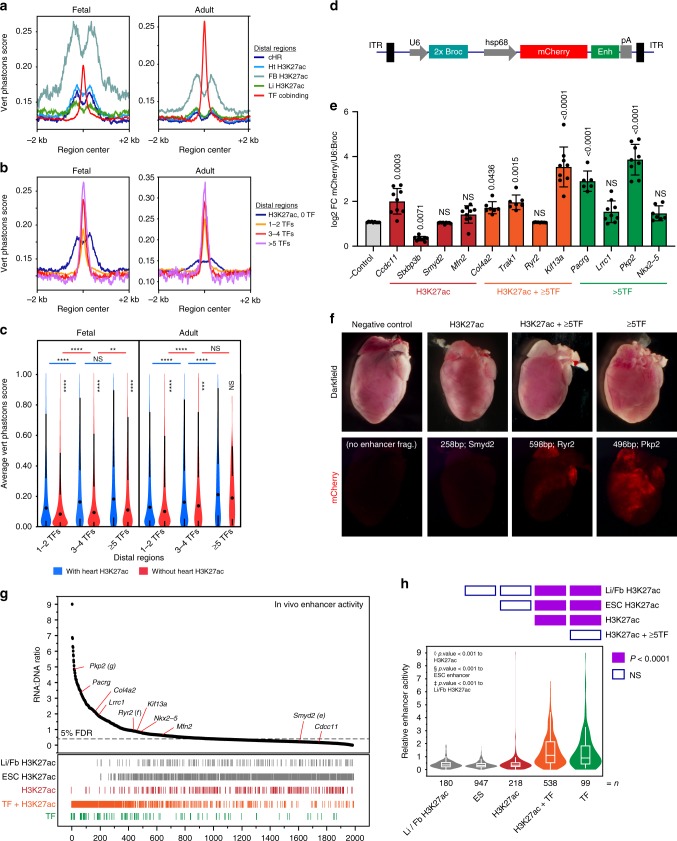
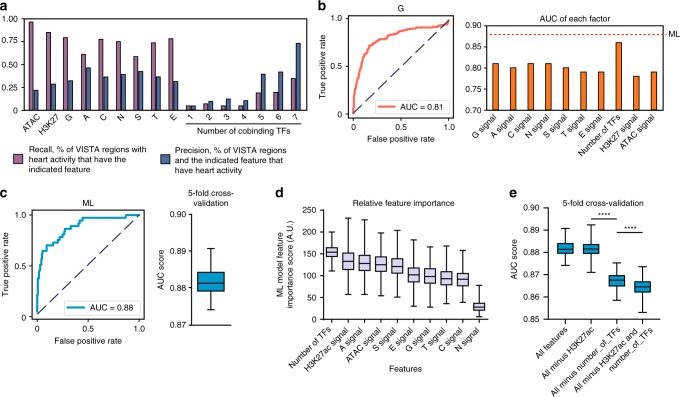
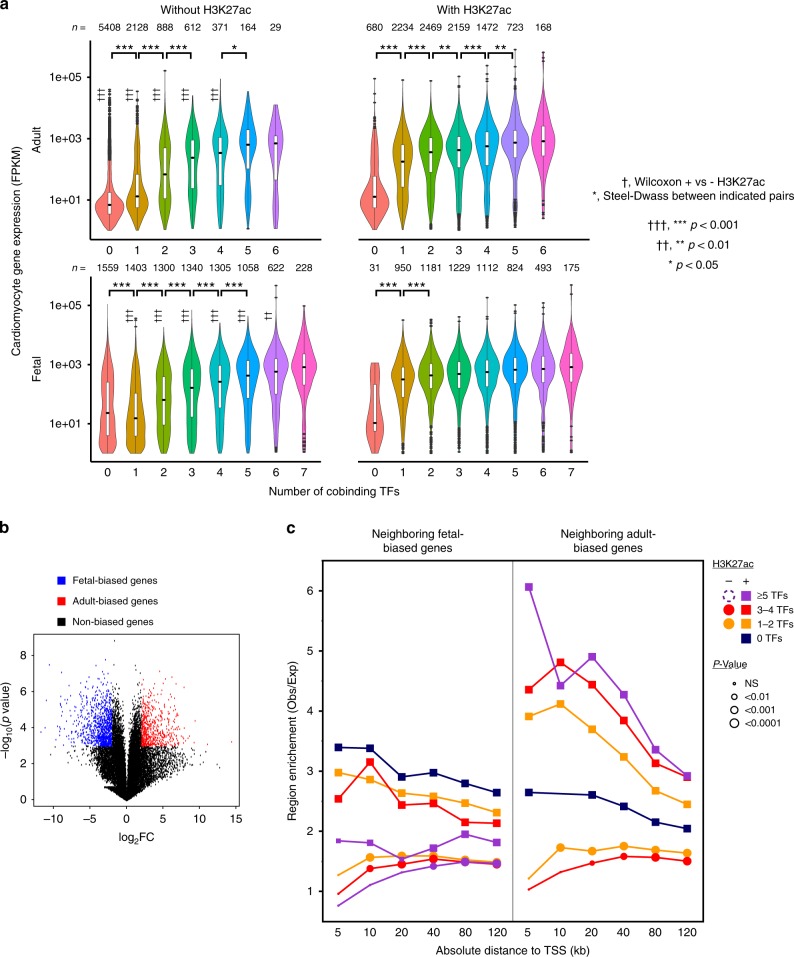
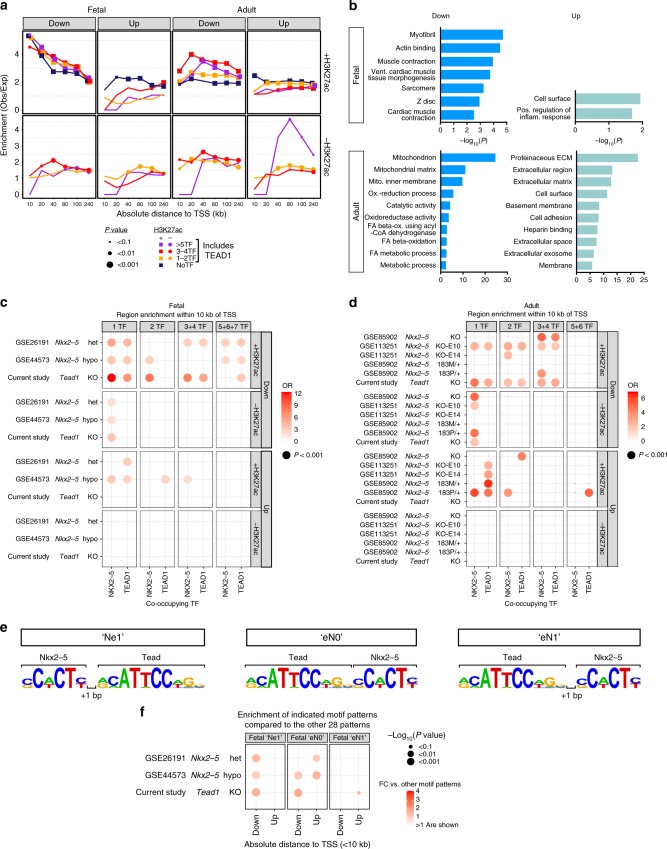
Mapping the chromatin occupancy of transcription factors (TFs) is a key step in deciphering developmental transcriptional programs. Here we use biotinylated knockin alleles of seven key cardiac TFs (GATA4, NKX2-5, MEF2A, MEF2C, SRF, TBX5, TEAD1) to sensitively and reproducibly map their genome-wide occupancy in the fetal and adult mouse heart. These maps show that TF occupancy is dynamic between developmental stages and that multiple TFs often collaboratively occupy the same chromatin region through indirect cooperativity. Multi-TF regions exhibit features of functional regulatory elements, including evolutionary conservation, chromatin accessibility, and activity in transcriptional enhancer assays. H3K27ac, a feature of many enhancers, incompletely overlaps multi-TF regions, and multi-TF regions lacking H3K27ac retain conservation and enhancer activity. TEAD1 is a core component of the cardiac transcriptional network, co-occupying cardiac regulatory regions and controlling cardiomyocyte-specific gene functions. Our study provides a resource for deciphering the cardiac transcriptional regulatory network and gaining insights into the molecular mechanisms governing heart development.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
