

Tumor cells hijack enteric glia to activate colon cancer stem cells and stimulate tumorigenesis
- PMID: 31662289
- PMCID: PMC6945247
- DOI: 10.1016/j.ebiom.2019.09.045
Tumor cells hijack enteric glia to activate colon cancer stem cells and stimulate tumorigenesis
Erratum in
-
Erratum to "Tumor cells hijack enteric glia to activate colon cancer stem cells and stimulate tumorigenesis" [EBioMedicine 49 (2019) 172-188].EBioMedicine. 2023 Feb;88:104448. doi: 10.1016/j.ebiom.2023.104448. Epub 2023 Jan 21. EBioMedicine. 2023. PMID: 36689913 Free PMC article. No abstract available.
Abstract
Background: Colon cancer stem cells (CSCs), considered responsible for tumor initiation and cancer relapse, are constantly exposed to regulatory cues emanating from neighboring cells present in the tumor microenvironment. Among these cells are enteric glial cells (EGCs) that are potent regulators of the epithelium functions in a healthy intestine. However, whether EGCs impact CSC-driven tumorigenesis remains unknown.
Methods: Impact of human EGC primary cultures or a non-transformed EGC line on CSCs isolated from human primary colon adenocarcinomas or colon cancer cell lines with different p53, MMR system and stemness status was determined using murine xenograft models and 3D co-culture systems. Supernatants of patient-matched human primary colon adenocarcinomas and non-adjacent healthy mucosa were used to mimic tumor versus healthy mucosa secretomes and compare their effects on EGCs.
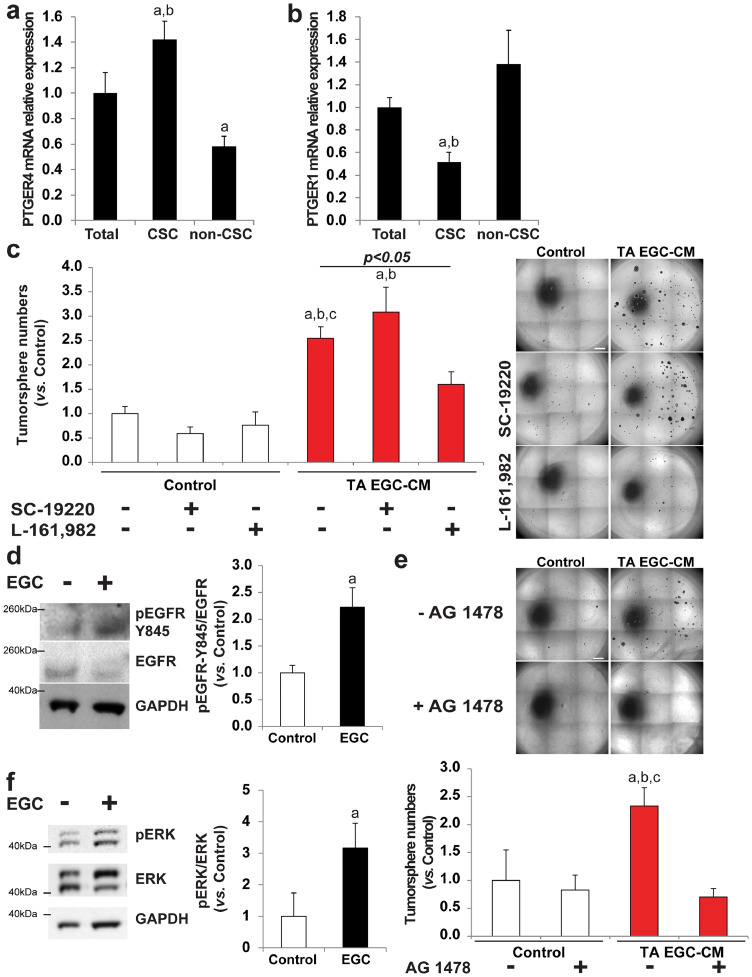
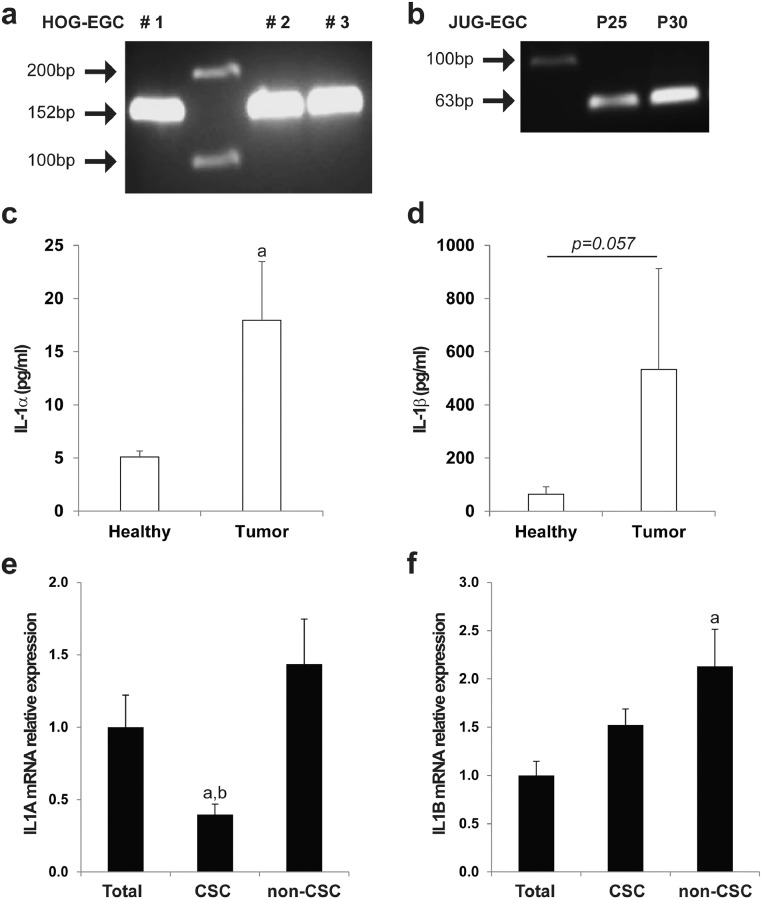
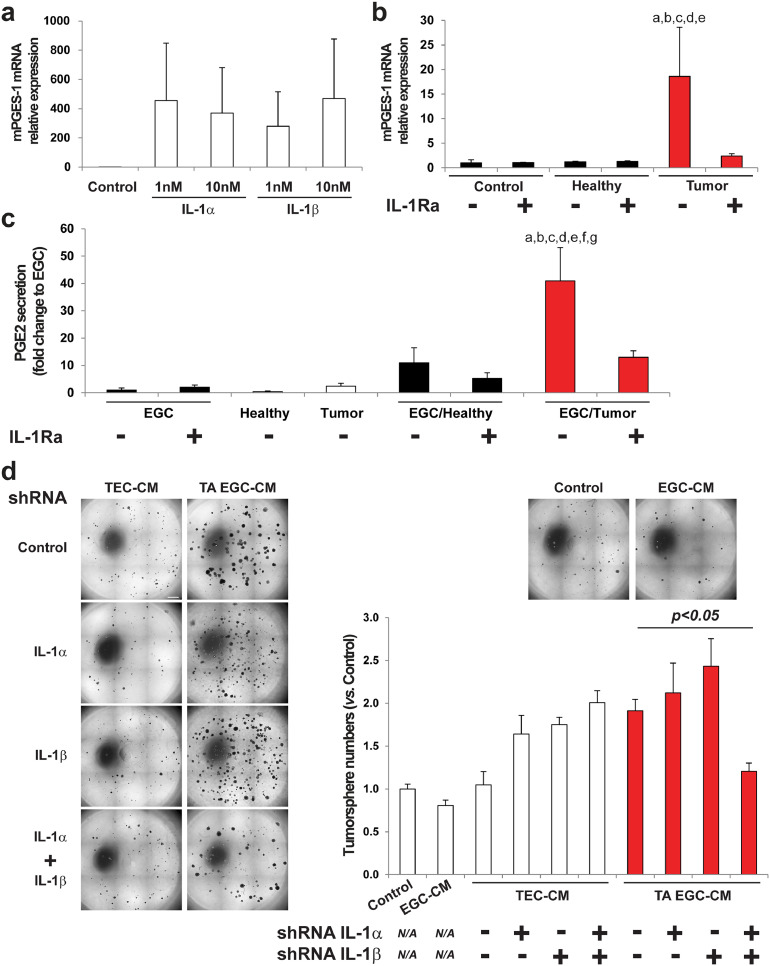
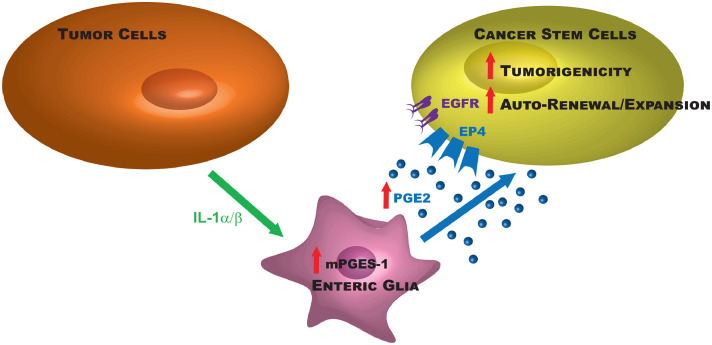
Findings: Our data show that EGCs stimulate CSC expansion and ability to give rise to tumors via paracrine signaling. Importantly, only EGCs that were pre-activated by tumor epithelial cell-derived soluble factors increased CSC tumorigenicity. Pharmacological inhibition of PGE2 biosynthesis in EGCs or IL-1 knockdown in tumor epithelial cells prevented EGC acquisition of a pro-tumorigenic phenotype. Inhibition of PGE2 receptor EP4 and EGFR in CSCs inhibited the effects of tumor-activated EGCs.
Interpretation: Altogether, our results show that EGCs, once activated by the tumor, acquire a pro-tumorigenic phenotype and stimulate CSC-driven tumorigenesis via a PGE2/EP4/EGFR-dependent pathway.
Funding: This work was supported by grants from the French National Cancer Institute, La Ligue contre le Cancer, the 'Région des Pays de la Loire' and the UNC Lineberger Comprehensive Cancer Center.
Keywords: Colon cancer stem cells; Colorectal cancer: Tumor microenvironment; Enteric glial cells; PGE2.
Copyright © 2019 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
All the authors declare no potential conflict of interest.
Figures
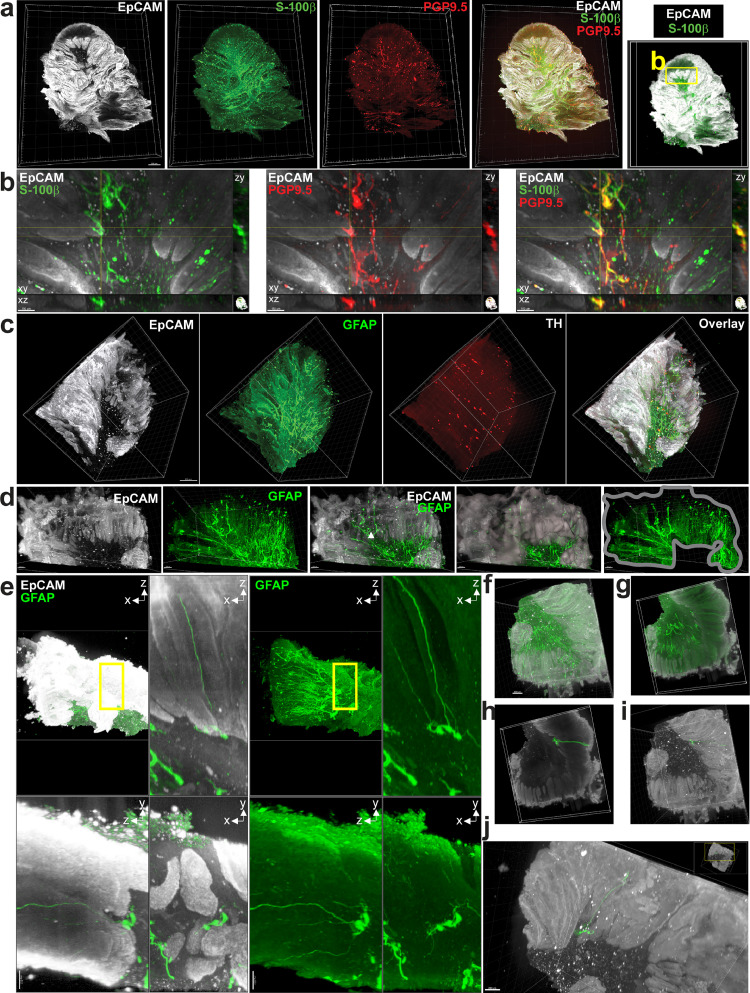
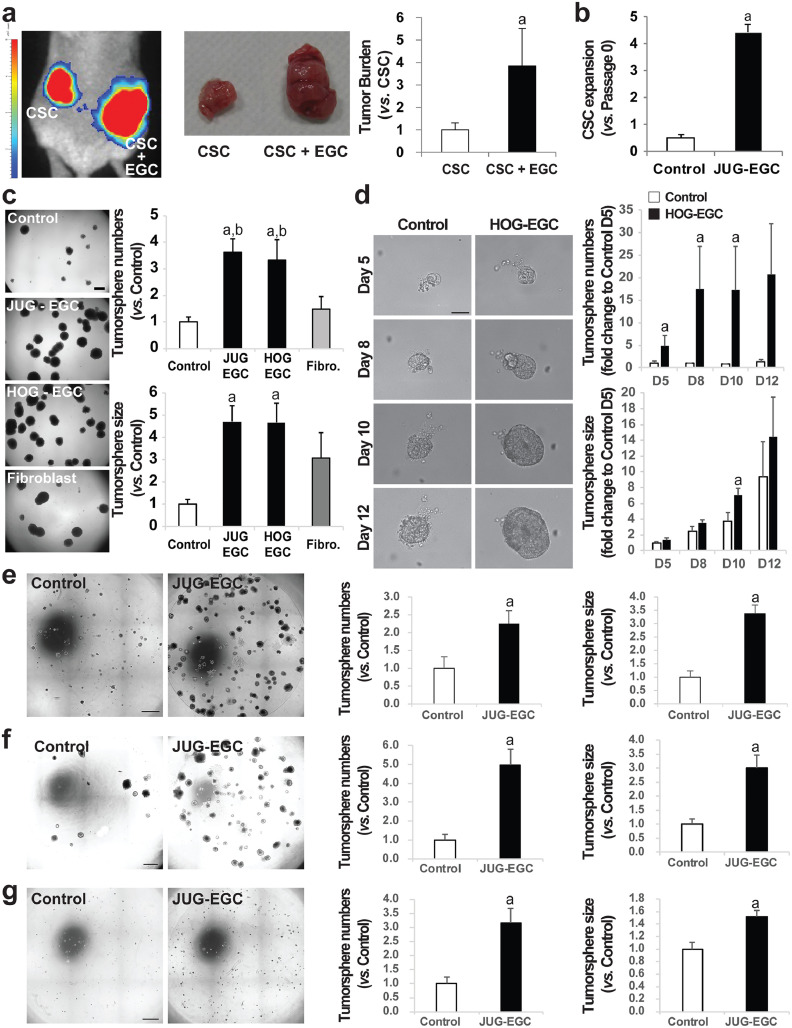
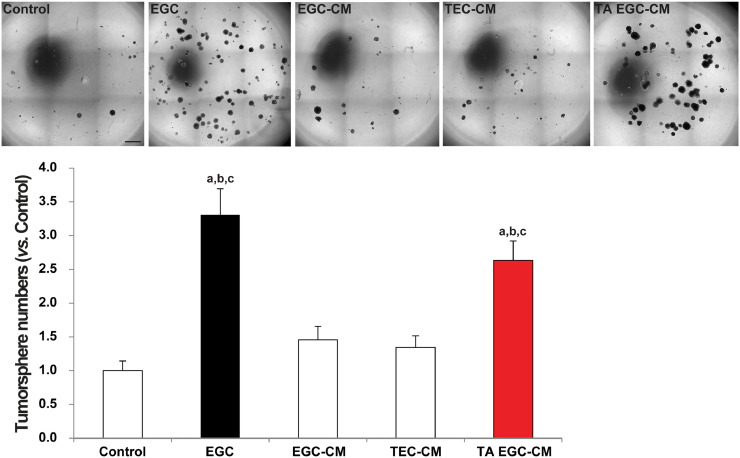
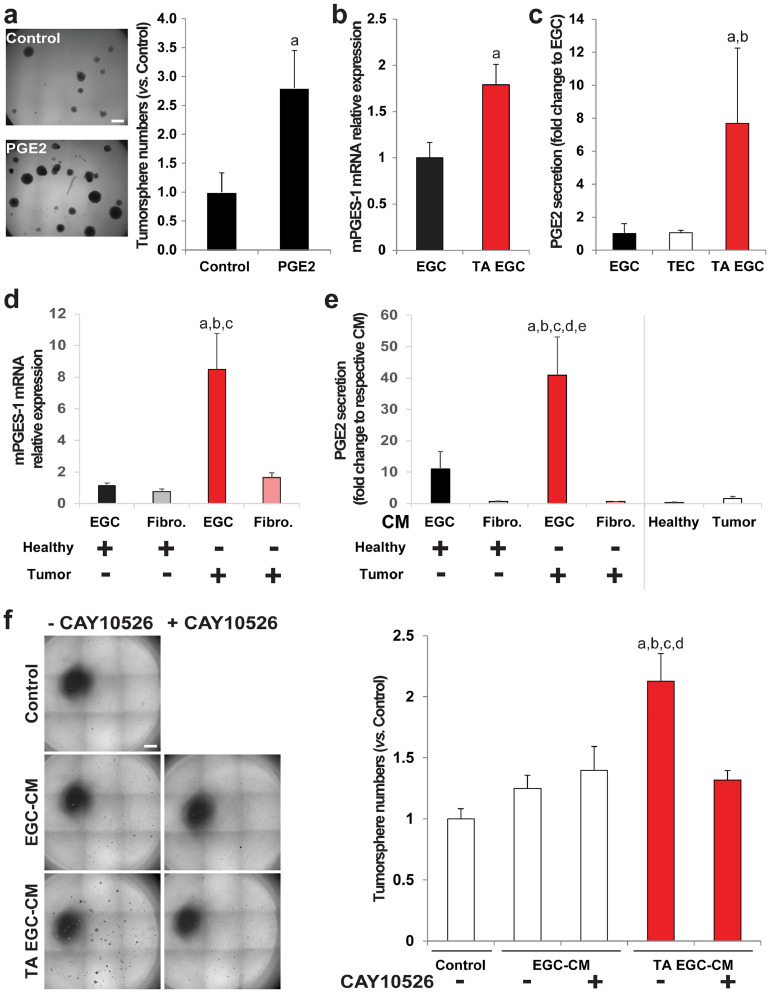
References
-
- Ricci-Vitiani L., Lombardi D.G., Pilozzi E., Biffoni M., Todaro M., Peschle C. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007 4;445(7123):111–115. - PubMed
-
- O'Brien C.A., Pollett A., Gallinger S., Dick J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007 4;445(7123):106–110. - PubMed
-
- Gao W., Chen L., Ma Z., Du Z., Zhao Z., Hu Z. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology. 2013;145(3):636–646. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
