

Chloroquine modulates inflammatory autoimmune responses through Nurr1 in autoimmune diseases
- PMID: 31664129
- PMCID: PMC6820774
- DOI: 10.1038/s41598-019-52085-w
Chloroquine modulates inflammatory autoimmune responses through Nurr1 in autoimmune diseases
Abstract
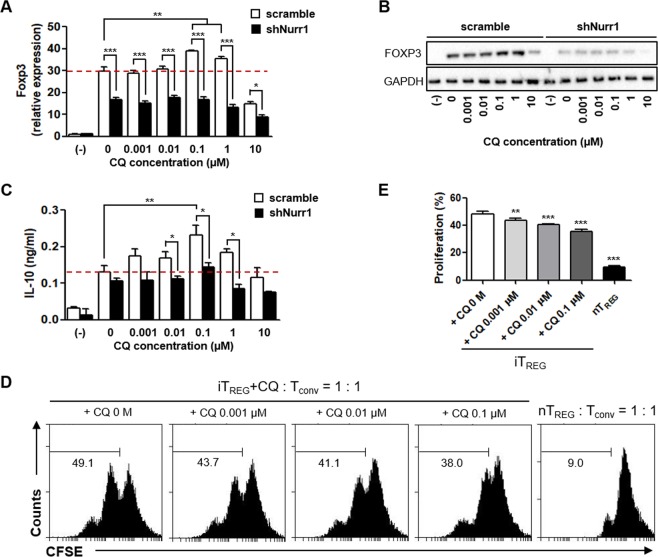
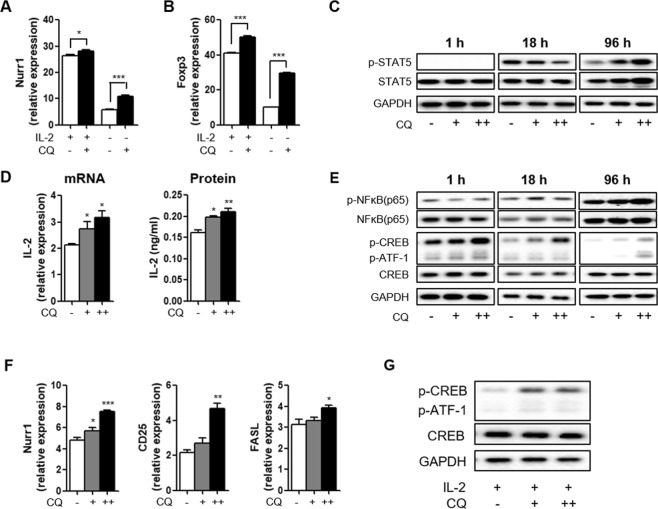
For over a half-century the anti-malarial drug chloroquine (CQ) has been used as a therapeutic agent, alone or in combination, to treat autoimmune diseases. However, neither the underlying mechanism(s) of action nor their molecular target(s) are well defined. The orphan nuclear receptor Nurr1 (also known as NR4A2) is an essential transcription factor affecting the development and maintenance of midbrain dopaminergic neurons. In this study, using in vitro T cell differentiation models, we demonstrate that CQ activates TREG cell differentiation and induces Foxp3 gene expression in a Nurr1-dependent manner. Remarkably, CQ appears to induce Nurr1 function by two distinct mechanisms: firstly, by direct binding to Nurr1's ligand-binding domain and promoting its transcriptional activity and secondly by upregulation of Nurr1 expression through the CREB signaling pathway. In contrast, CQ suppressed gene expression and differentiation of pathogenic TH17 cells. Importantly, using a valid animal model of inflammatory bowel disease (IBD), we demonstrated that CQ promotes Foxp3 expression and differentiation of TREG cells in a Nurr1-dependent manner, leading to significant improvement of IBD-related symptoms. Taken together, these data suggest that CQ ameliorates autoimmune diseases via regulating Nurr1 function/expression and that Nurr1 is a promising target for developing effective therapeutics of human inflammatory autoimmune diseases.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
- NS070577/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)/International
- NS084869/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)/International
- OD024622/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)/International
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
