

Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma
- PMID: 31664194
- PMCID: PMC7141958
- DOI: 10.1038/s41586-019-1650-0
Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma
Abstract
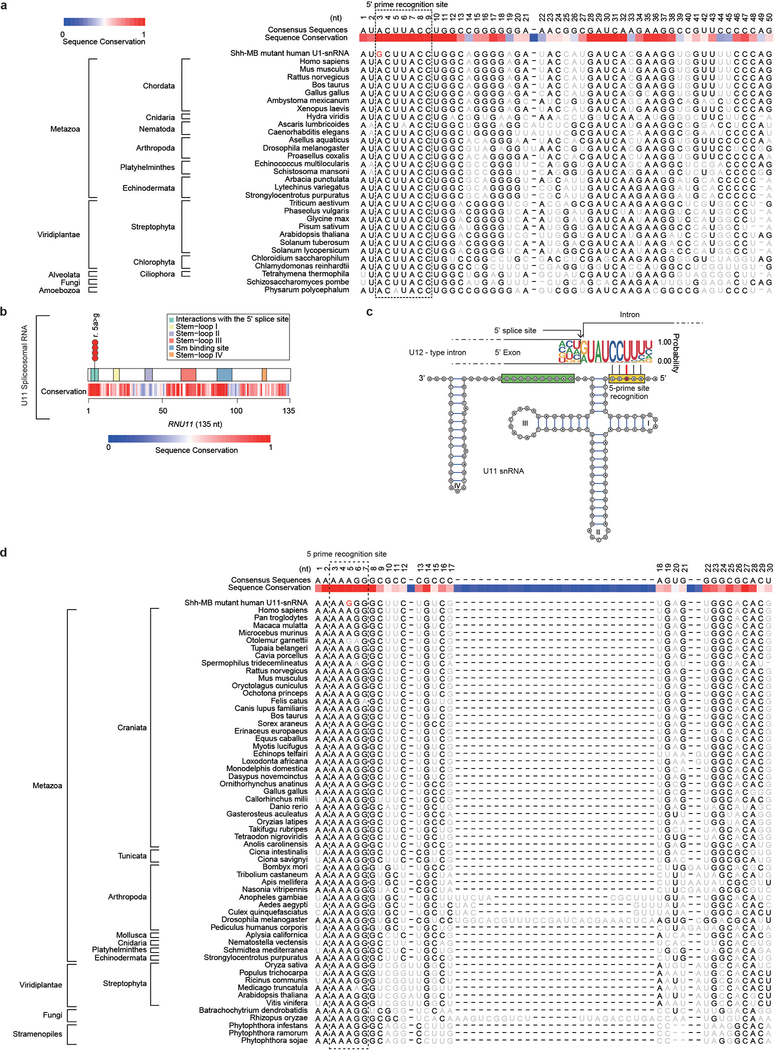
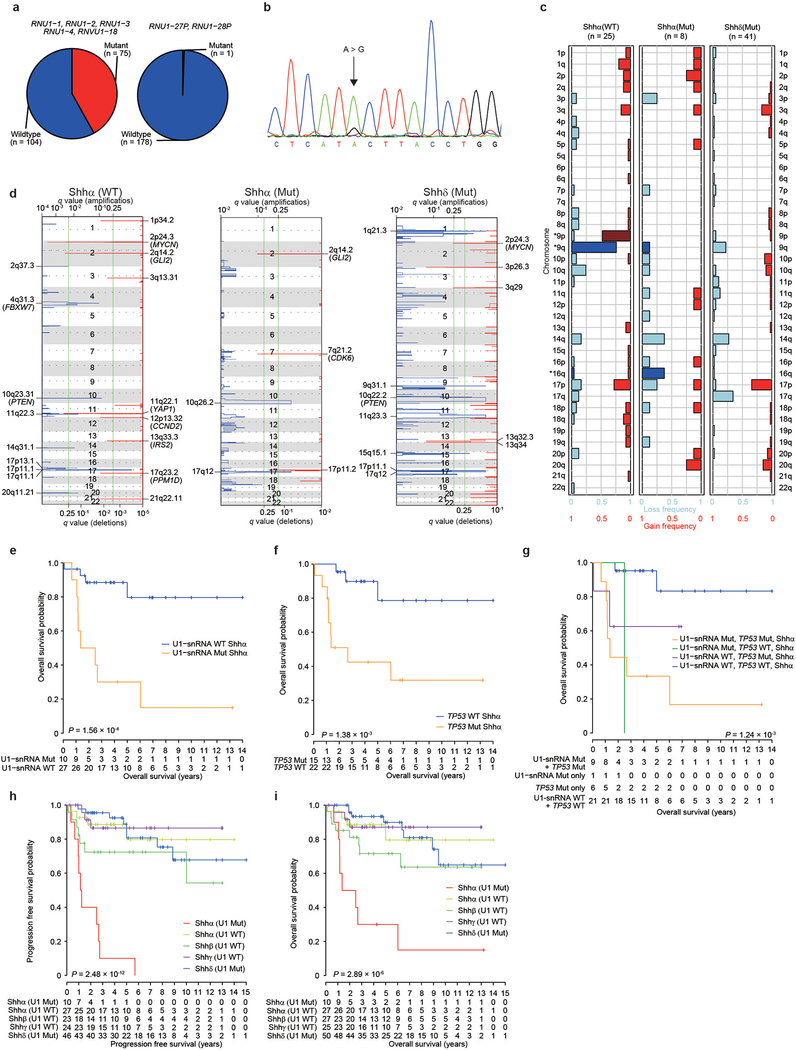
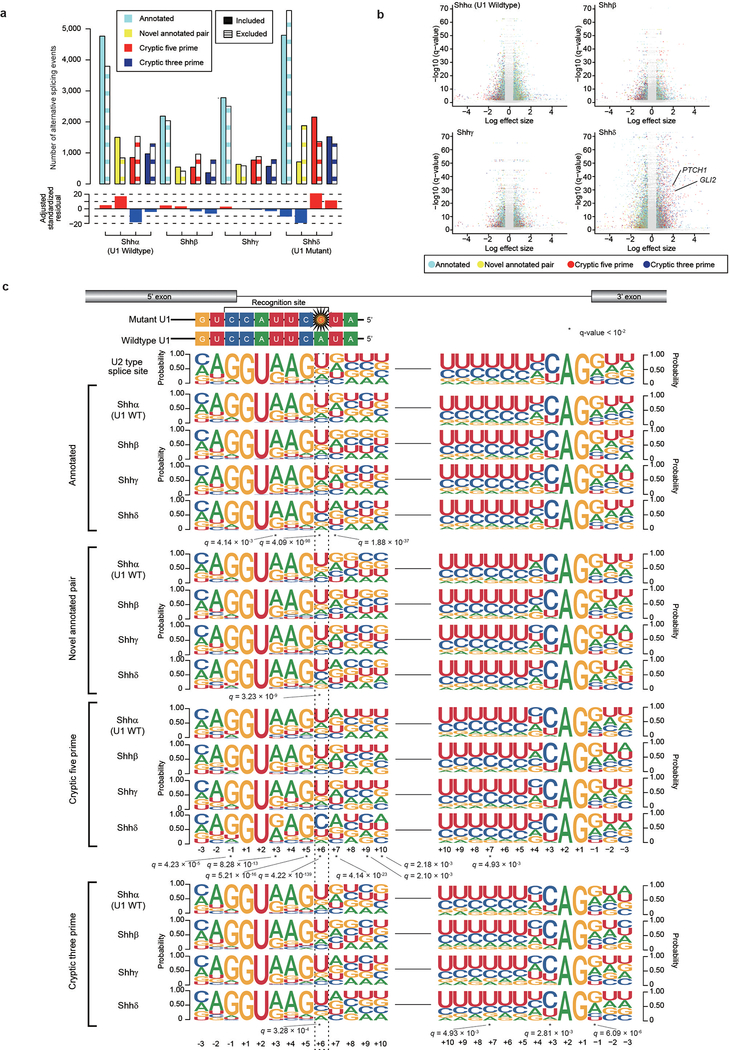
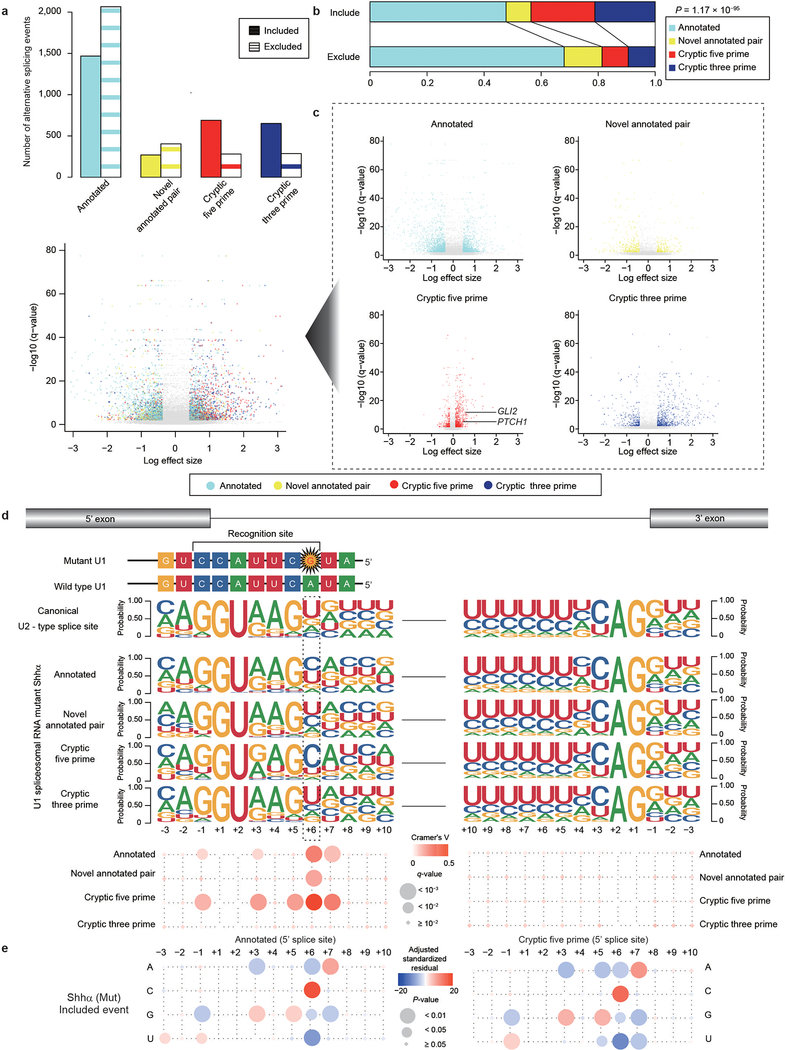
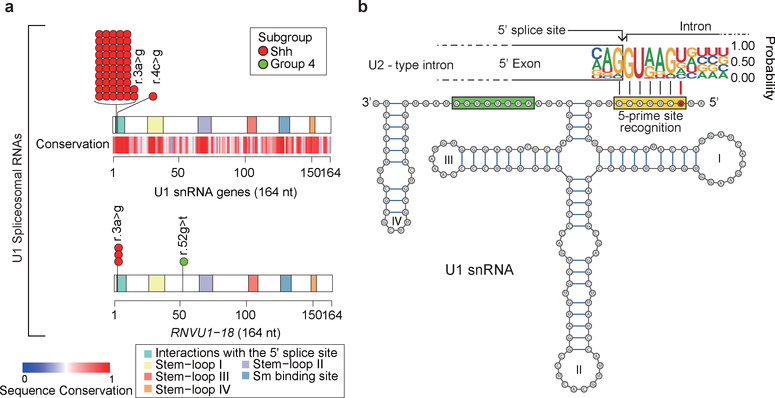
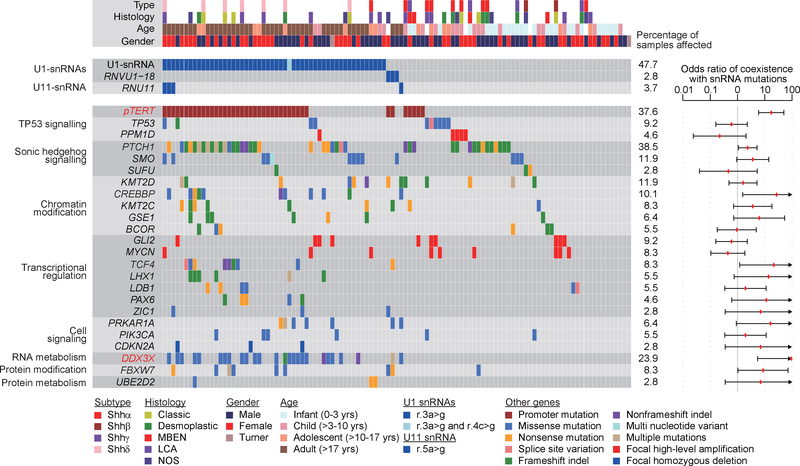
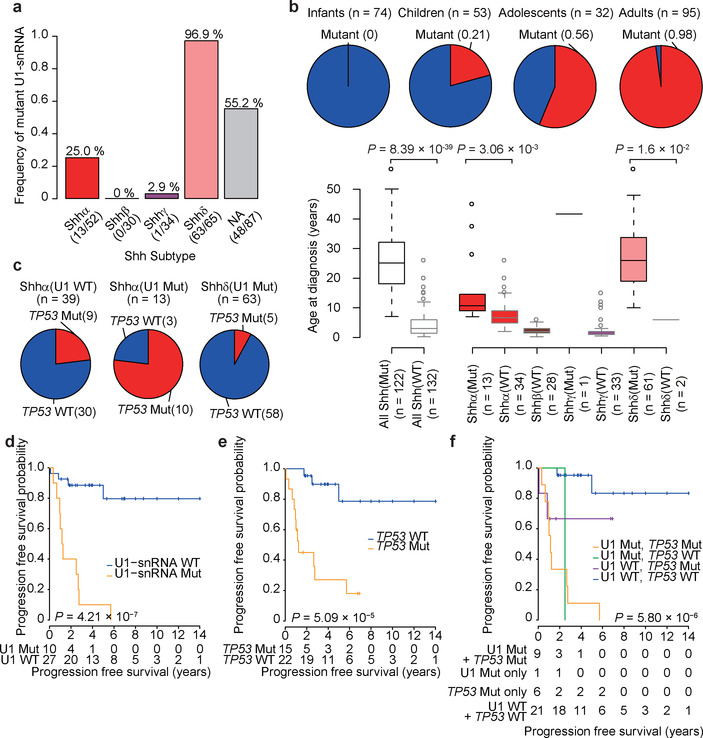
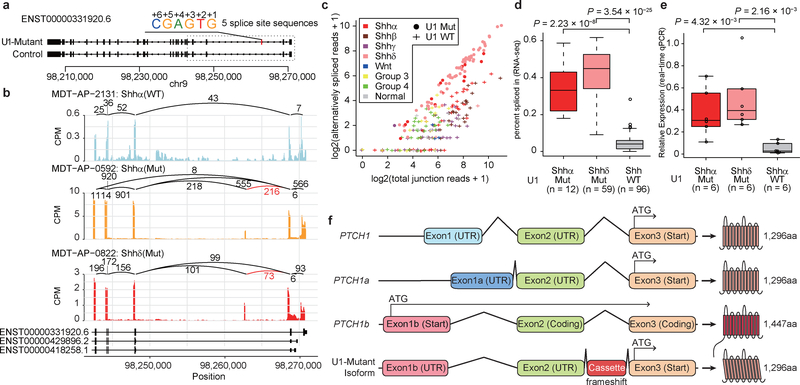
In cancer, recurrent somatic single-nucleotide variants-which are rare in most paediatric cancers-are confined largely to protein-coding genes1-3. Here we report highly recurrent hotspot mutations (r.3A>G) of U1 spliceosomal small nuclear RNAs (snRNAs) in about 50% of Sonic hedgehog (SHH) medulloblastomas. These mutations were not present across other subgroups of medulloblastoma, and we identified these hotspot mutations in U1 snRNA in only <0.1% of 2,442 cancers, across 36 other tumour types. The mutations occur in 97% of adults (subtype SHHδ) and 25% of adolescents (subtype SHHα) with SHH medulloblastoma, but are largely absent from SHH medulloblastoma in infants. The U1 snRNA mutations occur in the 5' splice-site binding region, and snRNA-mutant tumours have significantly disrupted RNA splicing and an excess of 5' cryptic splicing events. Alternative splicing mediated by mutant U1 snRNA inactivates tumour-suppressor genes (PTCH1) and activates oncogenes (GLI2 and CCND2), and represents a target for therapy. These U1 snRNA mutations provide an example of highly recurrent and tissue-specific mutations of a non-protein-coding gene in cancer.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures


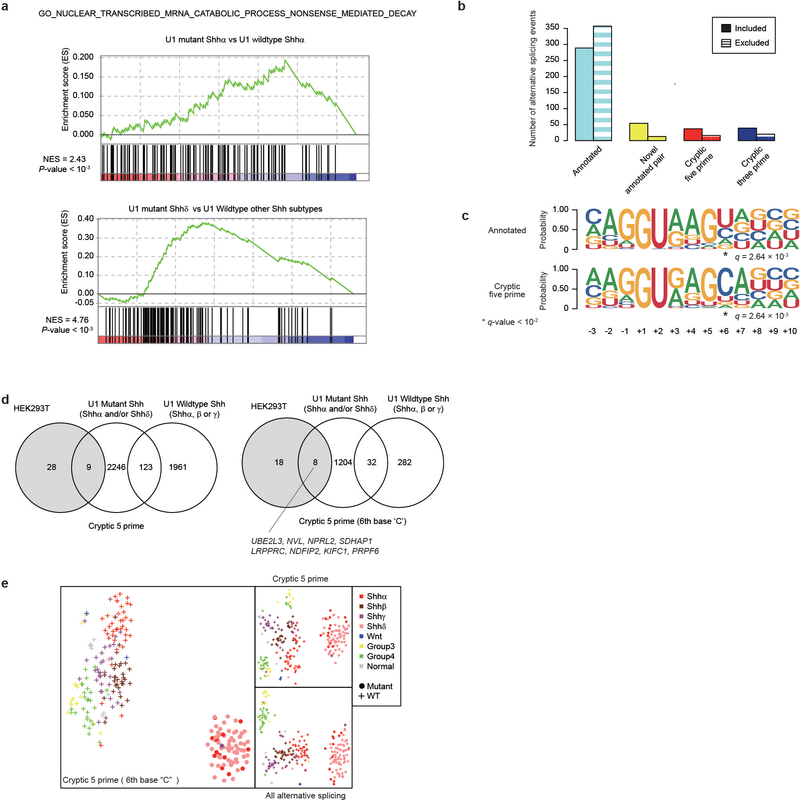
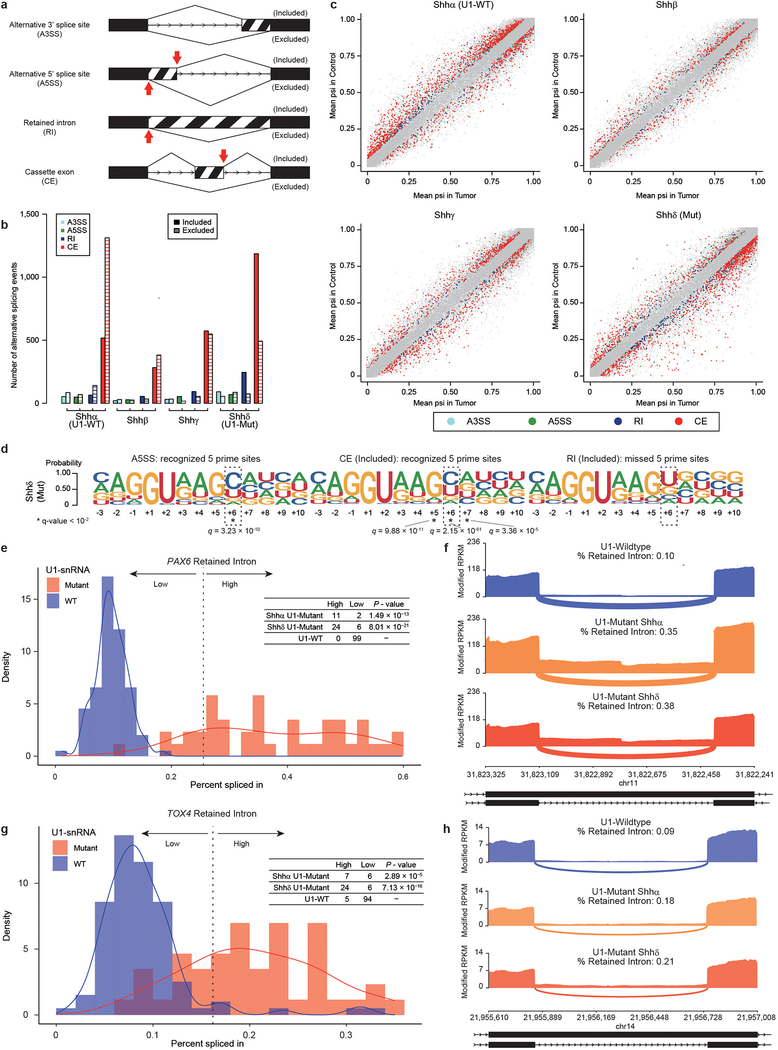
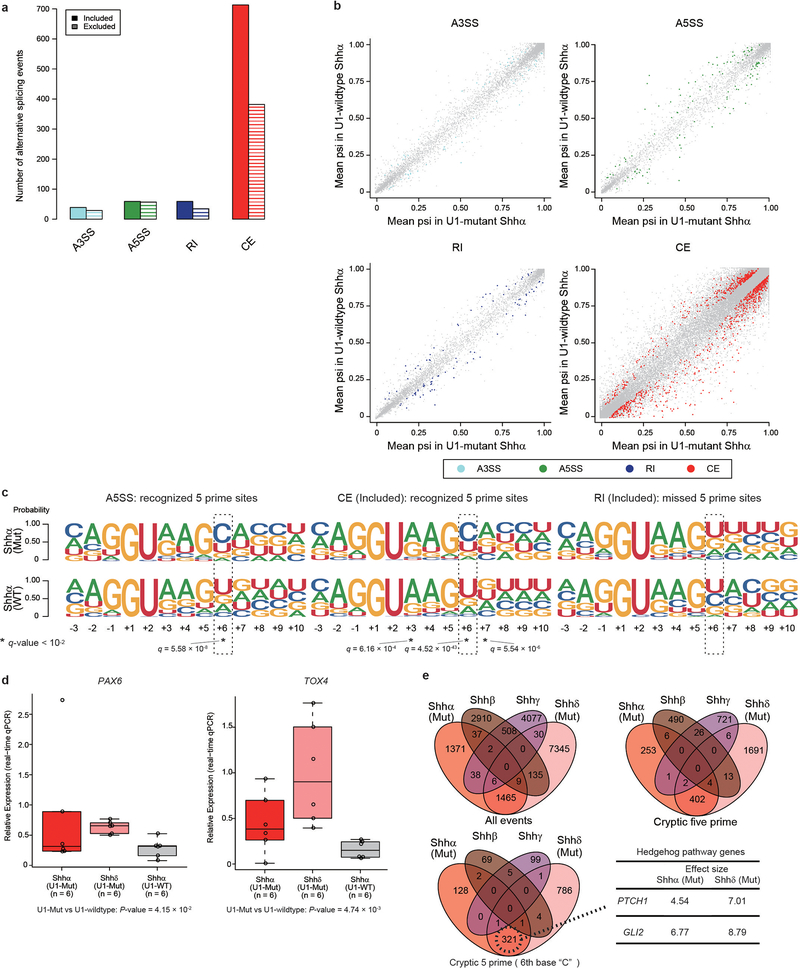
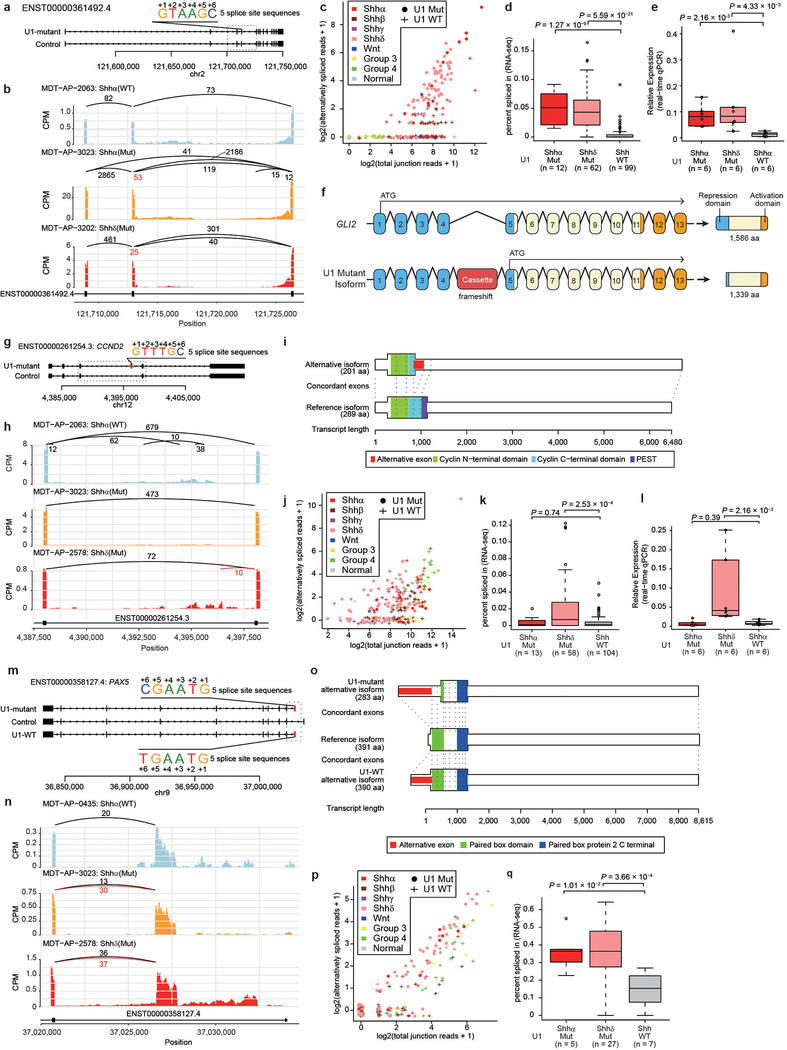
References
Methods References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
