

Bartter and Gitelman syndromes: Questions of class
- PMID: 31664557
- PMCID: PMC7501116
- DOI: 10.1007/s00467-019-04371-y
Bartter and Gitelman syndromes: Questions of class
Abstract
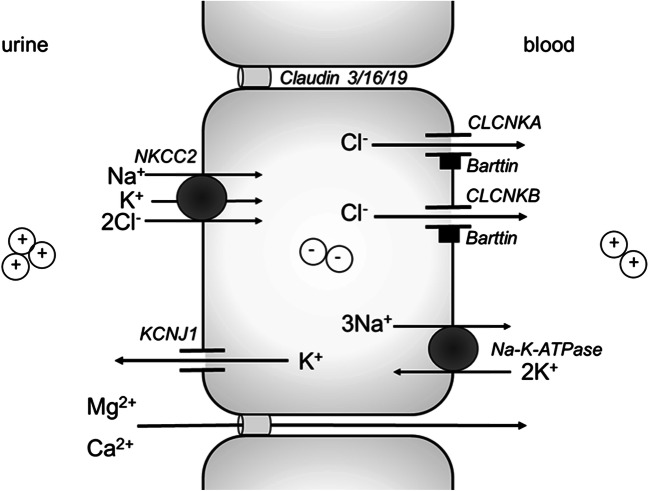
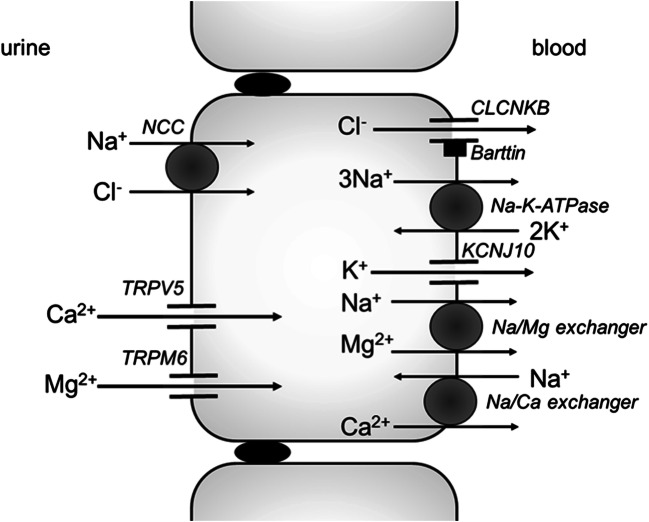
Bartter and Gitelman syndromes are rare inherited tubulopathies characterized by hypokalaemic, hypochloraemic metabolic alkalosis. They are caused by mutations in at least 7 genes involved in the reabsorption of sodium in the thick ascending limb (TAL) of the loop of Henle and/or the distal convoluted tubule (DCT). Different subtypes can be distinguished and various classifications have been proposed based on clinical symptoms and/or the underlying genetic cause. Yet, the clinical phenotype can show remarkable variability, leading to potential divergences between classifications. These problems mostly relate to uncertainties over the role of the basolateral chloride exit channel CLCNKB, expressed in both TAL and DCT and to what degree the closely related paralogue CLCNKA can compensate for the loss of CLCNKB function. Here, we review what is known about the physiology of the transport proteins involved in these disorders. We also review the various proposed classifications and explain why a gene-based classification constitutes a pragmatic solution.
Keywords: Bartter syndrome; EAST syndrome; Gitelman syndrome; Hypokalaemia; Metabolic alkalosis; Tubulopathy.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Madden N, Trachtman H. Physiology of the developing kidney: sodium and water homeostasis and its disorders. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein FL, editors. Pediatric Nephrology. Berlin: Springer-Verlag; 2016. pp. 181–217.
-
- Satlin LM, Bockenhauer D. Physiology of the developing kidney: potassium homeostasis and its disorder. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein FL, editors. Pediatric Nephrology. Berlin: Springer-Verlag; 2016. pp. 219–246.
-
- Seyberth HW, Weber S, Komhoff M. Bartterʼs and Gitelmanʼs syndrome. Curr Opin Pediatr. 2017;29(2):179–186. - PubMed
-
- Kramer BK, Bergler T, Stoelcker B, Waldegger S. Mechanisms of disease: the kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat Clin Pract Nephrol. 2008;4(1):38–46. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
