

Human mitochondrial DNA is extensively methylated in a non-CpG context
- PMID: 31665742
- PMCID: PMC6821263
- DOI: 10.1093/nar/gkz762
Human mitochondrial DNA is extensively methylated in a non-CpG context
Abstract
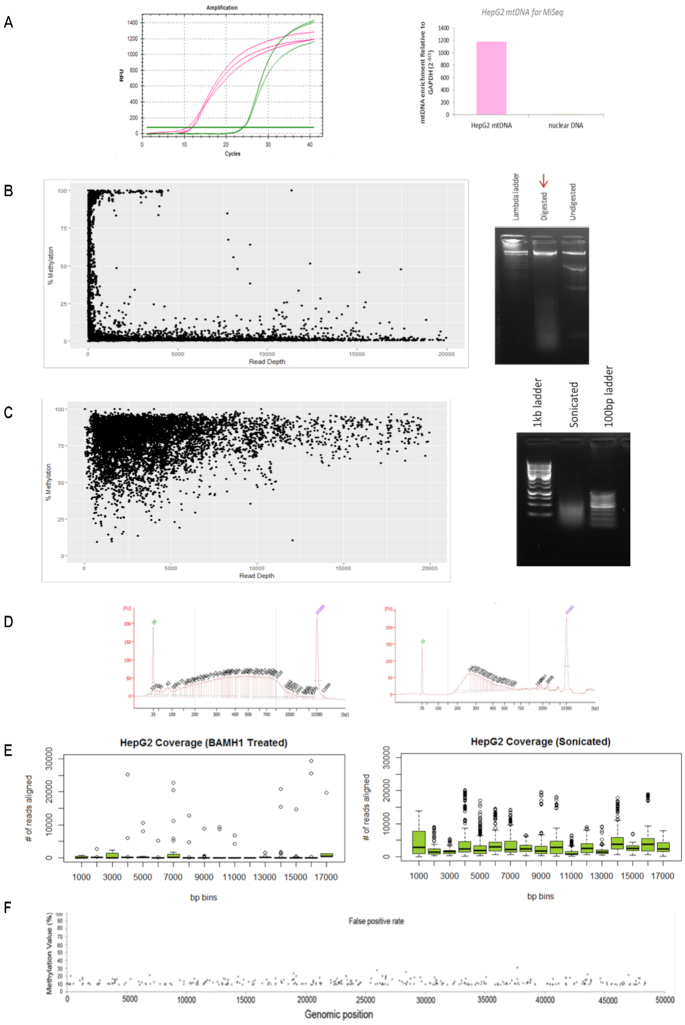
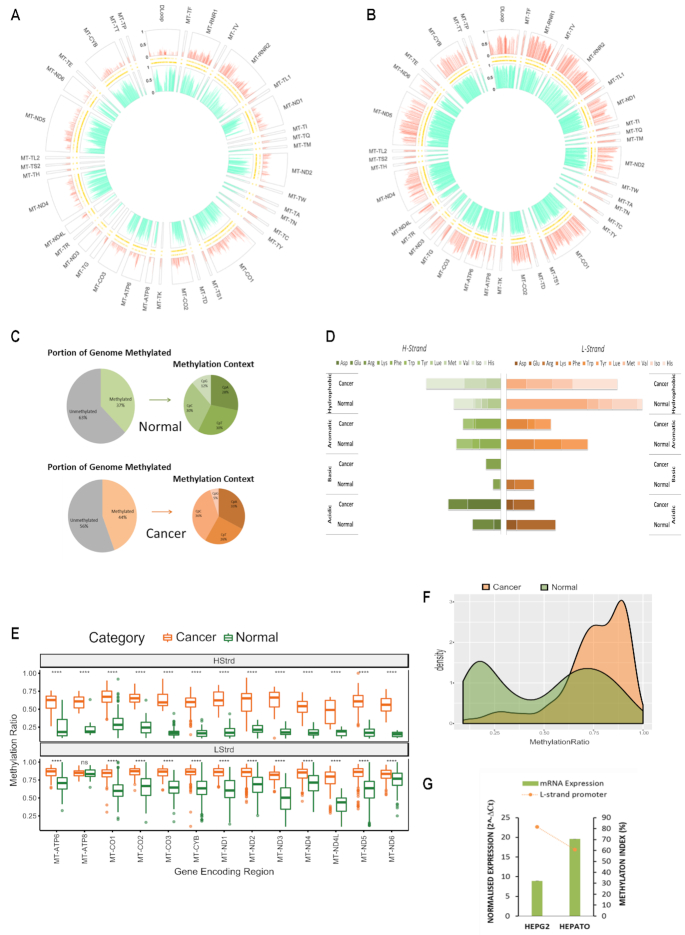
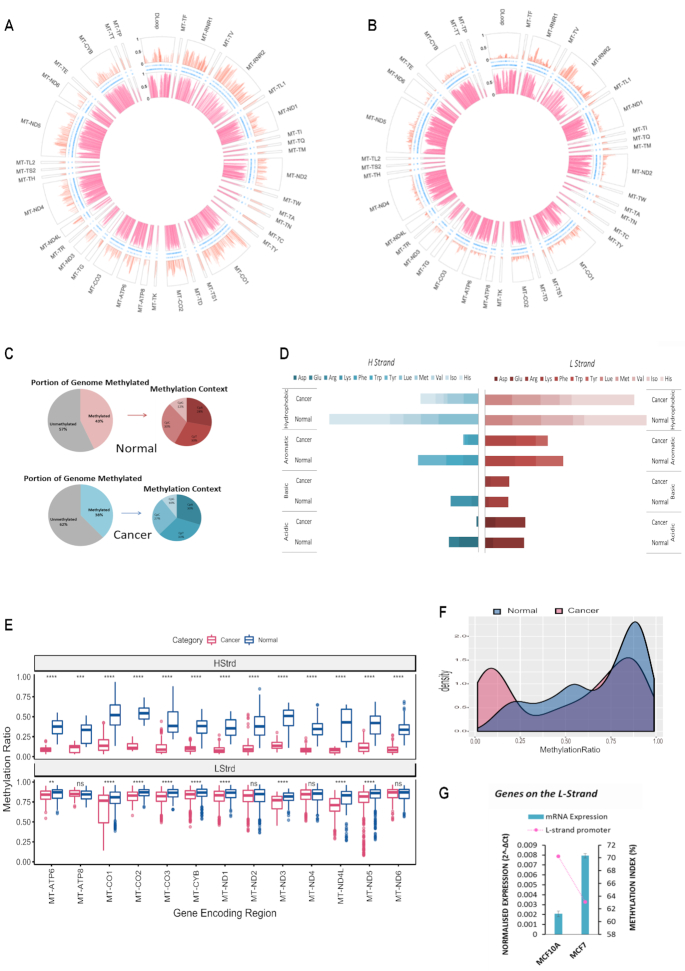
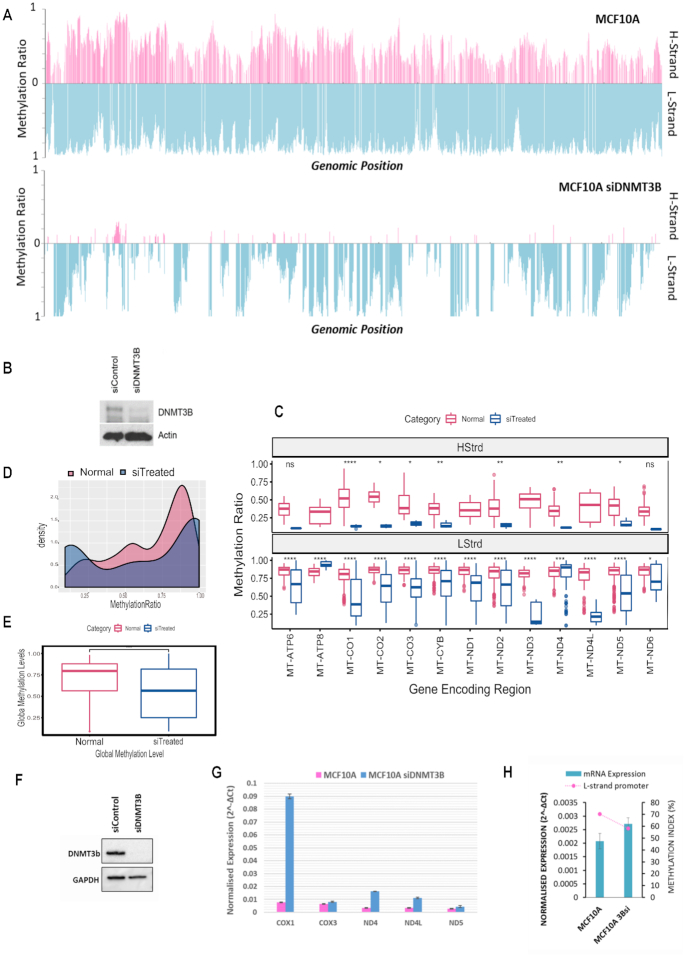
Mitochondrial dysfunction plays critical roles in cancer development and related therapeutic response; however, exact molecular mechanisms remain unclear. Recently, alongside the discovery of mitochondrial-specific DNA methyltransferases, global and site-specific methylation of the mitochondrial genome has been described. Investigation of any functional consequences however remains unclear and debated due to insufficient evidence of the quantitative degree and frequency of mitochondrial DNA (mtDNA) methylation. This study uses WGBS to provide the first quantitative report of mtDNA methylation at single base pair resolution. The data show that mitochondrial genomes are extensively methylated predominantly at non-CpG sites. Importantly, these methylation patterns display notable differences between normal and cancer cells. Furthermore, knockdown of DNA methyltransferase enzymes resulted in a marked global reduction of mtDNA methylation levels, indicating these enzymes may be associated with the establishment and/or maintenance of mtDNA methylation. DNMT3B knockdown cells displayed a comparatively pronounced global reduction in mtDNA methylation with concomitant increases in gene expression, suggesting a potential functional link between methylation and gene expression. Together these results demonstrate reproducible, non-random methylation patterns of mtDNA and challenge the notion that mtDNA is lowly methylated. This study discusses key differences in methodology that suggest future investigations must allow for techniques that assess both CpG and non-CpG methylation.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Tapper D.P., Clayton D.A.. Mechanism of replication of human mitochondrial DNA. Localization of the 5′ ends of nascent daughter strands. J. Biol. Chem. 1981; 256:5109–5115. - PubMed
-
- Crews S., Ojala D., Posakony J., Nishiguchi J., Attardi G.. Nucleotide sequence of a region of human mitochondrial DNA containing the precisely identified origin of replication. Nature. 1979; 277:192–198. - PubMed
-
- Fish J., Raule N., Attardi G.. Discovery of a major D-loop replication origin reveals two modes of human mtDNA synthesis. Science. 2004; 306:2098–2101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
