

An Improved Recombineering Toolset for Plants
- PMID: 31666295
- PMCID: PMC6961616
- DOI: 10.1105/tpc.19.00431
An Improved Recombineering Toolset for Plants
Abstract
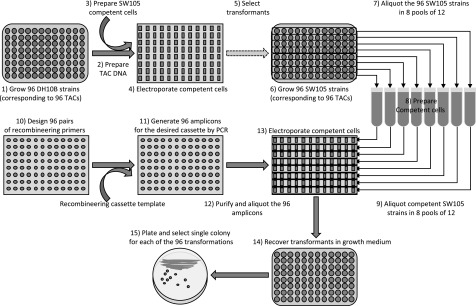
Gene functional studies often rely on the expression of a gene of interest as transcriptional and translational fusions with specialized tags. Ideally, this is done in the native chromosomal contexts to avoid potential misexpression artifacts. Although recent improvements in genome editing have made it possible to directly modify the target genes in their native chromosomal locations, classical transgenesis is still the preferred experimental approach chosen in most gene tagging studies because of its time efficiency and accessibility. We have developed a recombineering-based tagging system that brings together the convenience of the classical transgenic approaches and the high degree of confidence in the results obtained by direct chromosomal tagging using genome-editing strategies. These simple, scalable, customizable recombineering toolsets and protocols allow a variety of genetic modifications to be generated. In addition, we developed a highly efficient recombinase-mediated cassette exchange system to facilitate the transfer of the desired sequences from a bacterial artificial chromosome clone to a transformation-compatible binary vector, expanding the use of the recombineering approaches beyond Arabidopsis (Arabidopsis thaliana). We demonstrated the utility of this system by generating more than 250 whole-gene translational fusions and 123 Arabidopsis transgenic lines corresponding to 62 auxin-related genes and characterizing the translational reporter expression patterns for 14 auxin biosynthesis genes.
© 2020 American Society of Plant Biologists. All rights reserved.
Figures







References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
