

PAR2 controls cholesterol homeostasis and lipid metabolism in nonalcoholic fatty liver disease
- PMID: 31668396
- PMCID: PMC6742970
- DOI: 10.1016/j.molmet.2019.08.019
PAR2 controls cholesterol homeostasis and lipid metabolism in nonalcoholic fatty liver disease
Abstract
Objective: Increases in hepatic and plasma cholesterol occur in patients with nonalcoholic fatty liver disease (NAFLD), although the reason for this is not well understood. We investigated whether Protease-Activated Receptor 2 (PAR2) plays a role in cholesterol and lipid homeostasis in NAFLD.
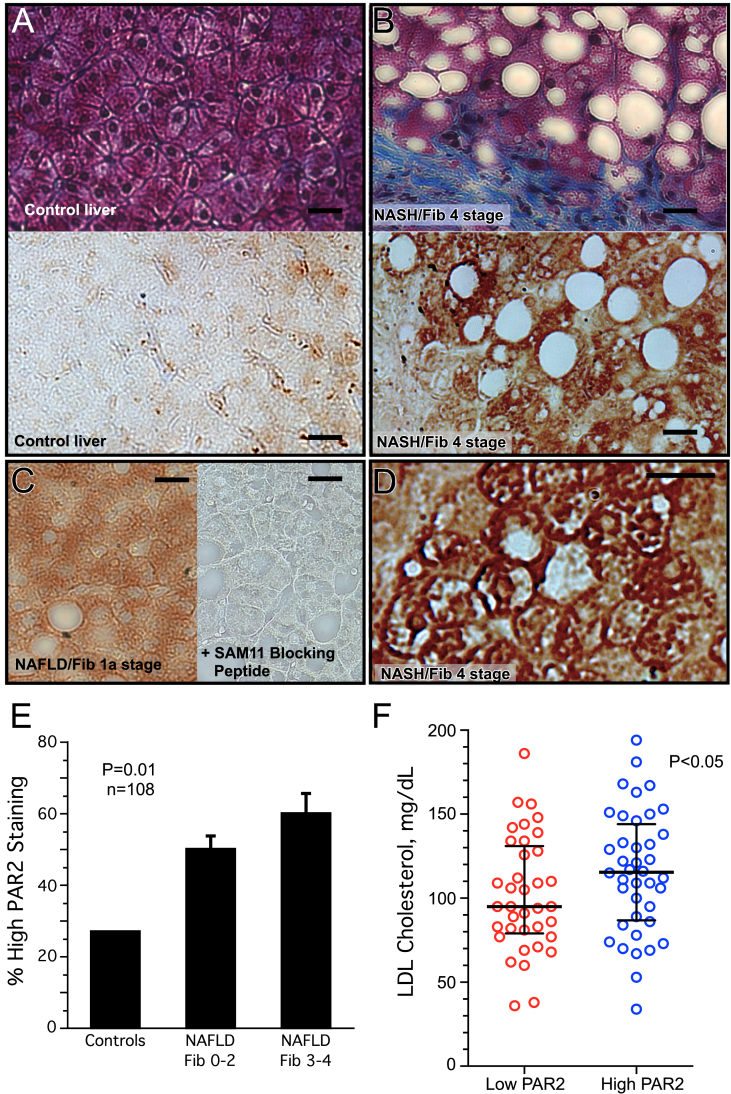
Methods: Human liver biopsies (n = 108) were quantified for PAR2 expression from NAFLD cases randomly selected and stratified by liver fibrosis stage, the primary predictor for clinical outcomes, while controlling for age, gender, and BMI between fibrosis groups. Demographic data and laboratory studies on plasma samples were obtained within 6 months of liver biopsy. Wild-type and PAR2-KO (C57BL/6 F2rl1-/-) mice were fed either normal or high fat diet for 16 weeks and plasma and liver assayed for lipids and soluble metabolites.
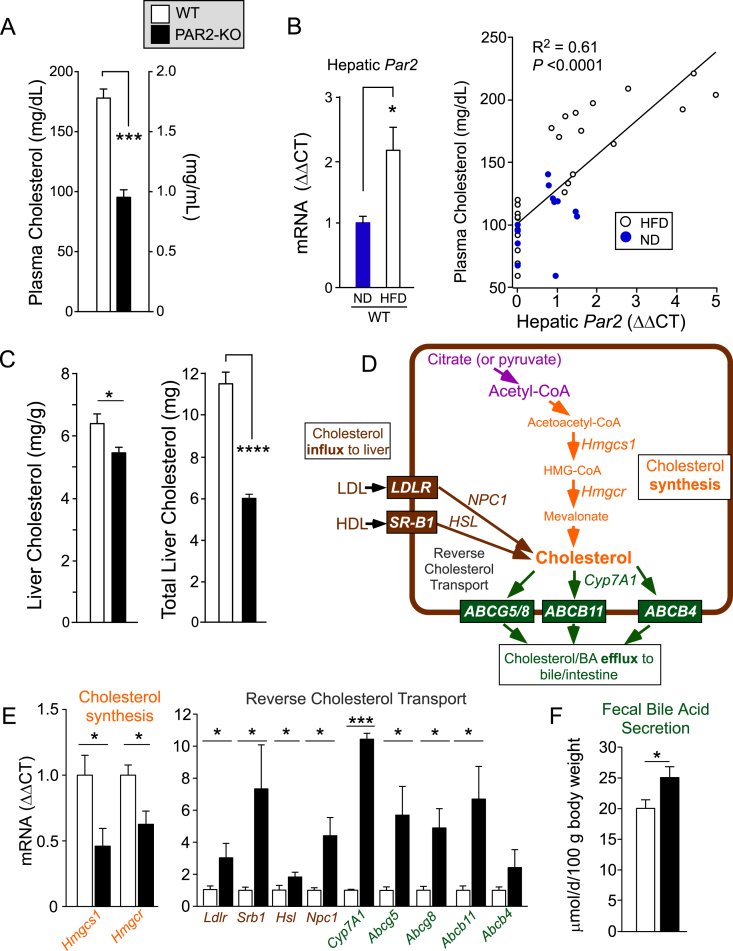
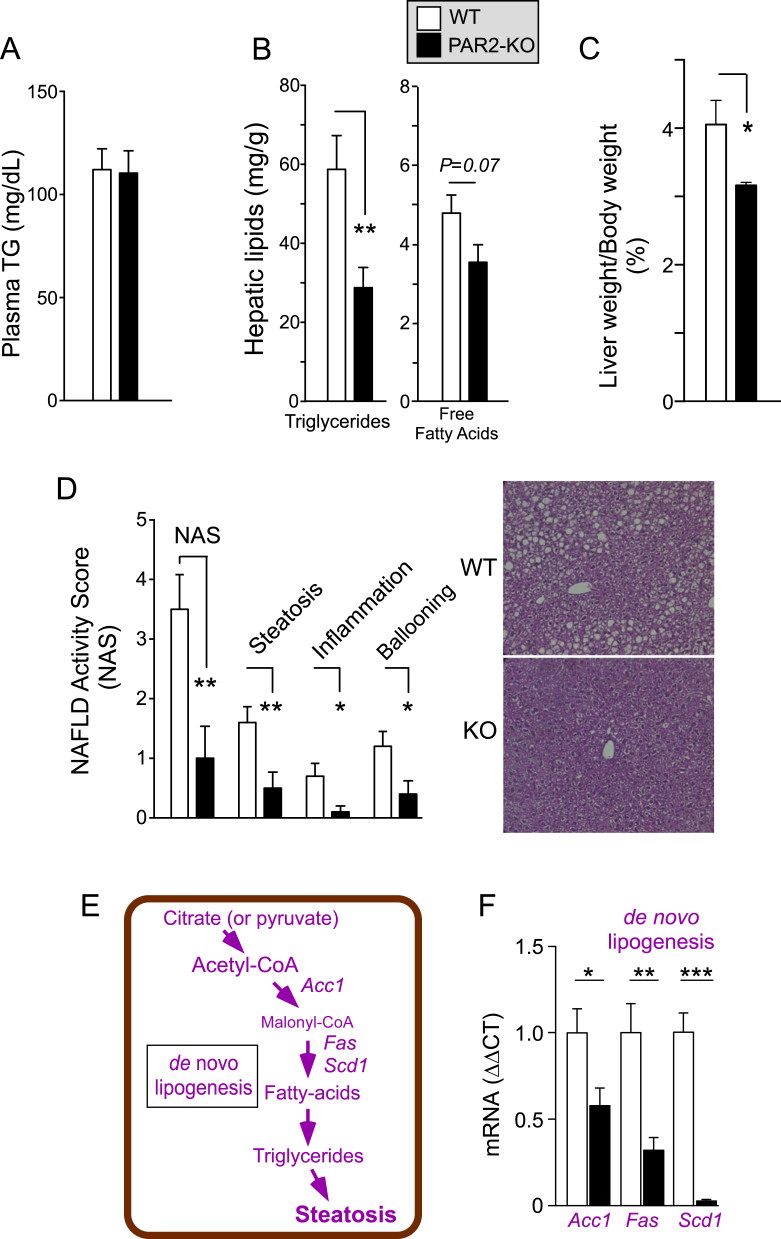
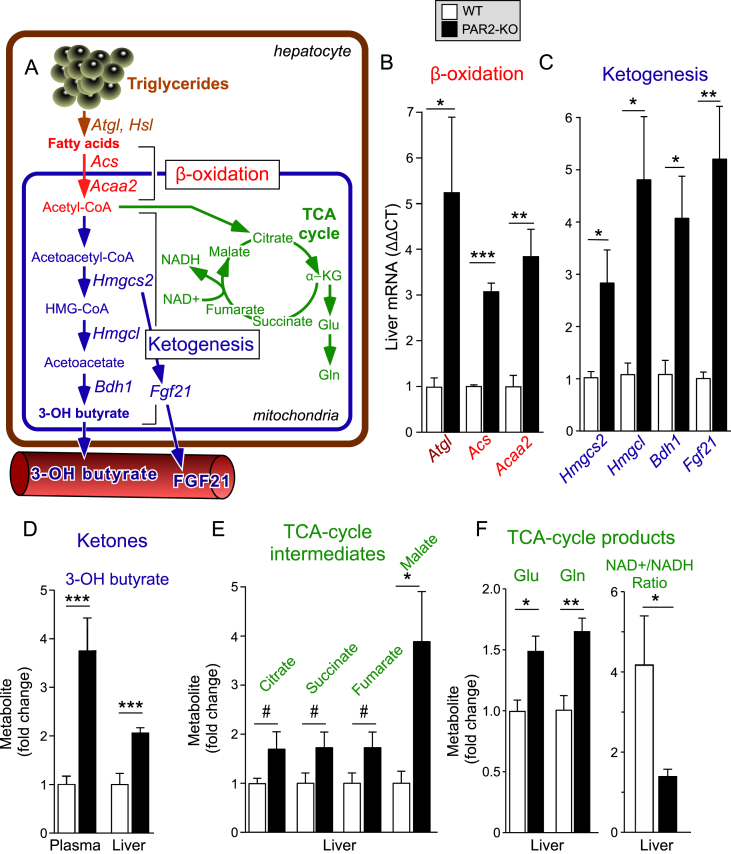
Results: Severity of NAFLD and plasma cholesterol levels significantly correlated with hepatocyte PAR2 expression in NAFLD patients. Conversely, PAR2 deficiency in mice resulted in reduced expression of key hepatic genes involved in cholesterol synthesis, a 50% drop in plasma and total liver cholesterol, and induced a reverse cholesterol transport system that culminated in 25% higher fecal bile acid output. PAR2-deficient mice exhibited enhanced fatty acid β-oxidation with a ketogenic shift and an unexpected increase in liver glycogenesis. Mechanistic studies identified Gi-Jnk1/2 as key downstream effectors of protease-activated PAR2 in the regulation of lipid and cholesterol homeostasis in liver.
Conclusions: These data indicate that PAR2 may be a new target for the suppression of plasma cholesterol and hepatic fat accumulation in NAFLD and related metabolic conditions.
Keywords: Cholesterol; Energy metabolism; Fatty liver; NASH; Protease-activated receptor 2.
Copyright © 2019 The Authors. Published by Elsevier GmbH.. All rights reserved.
Figures
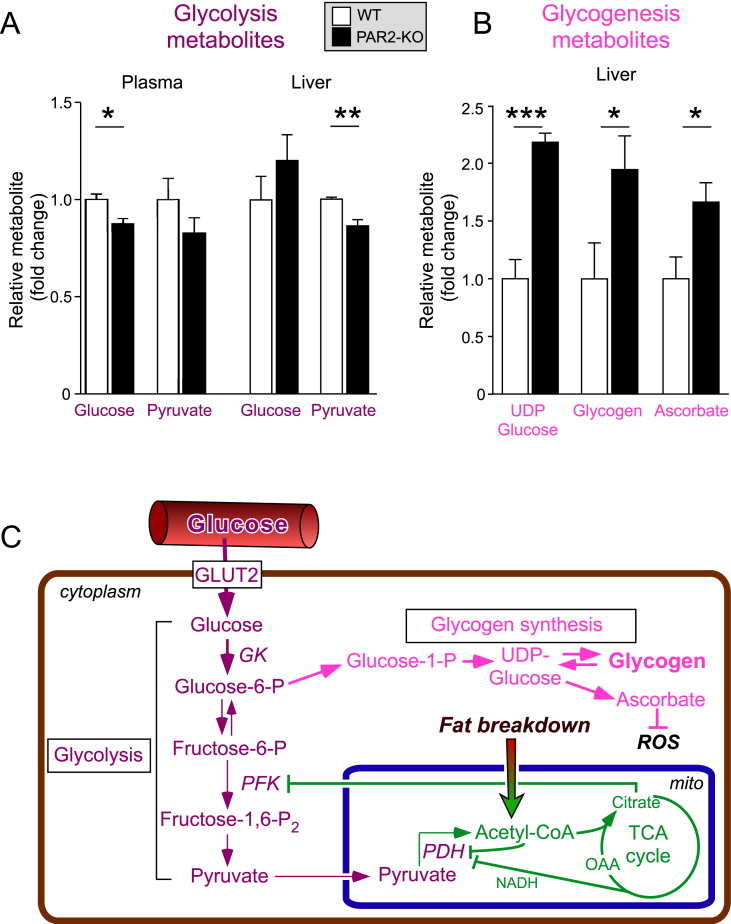
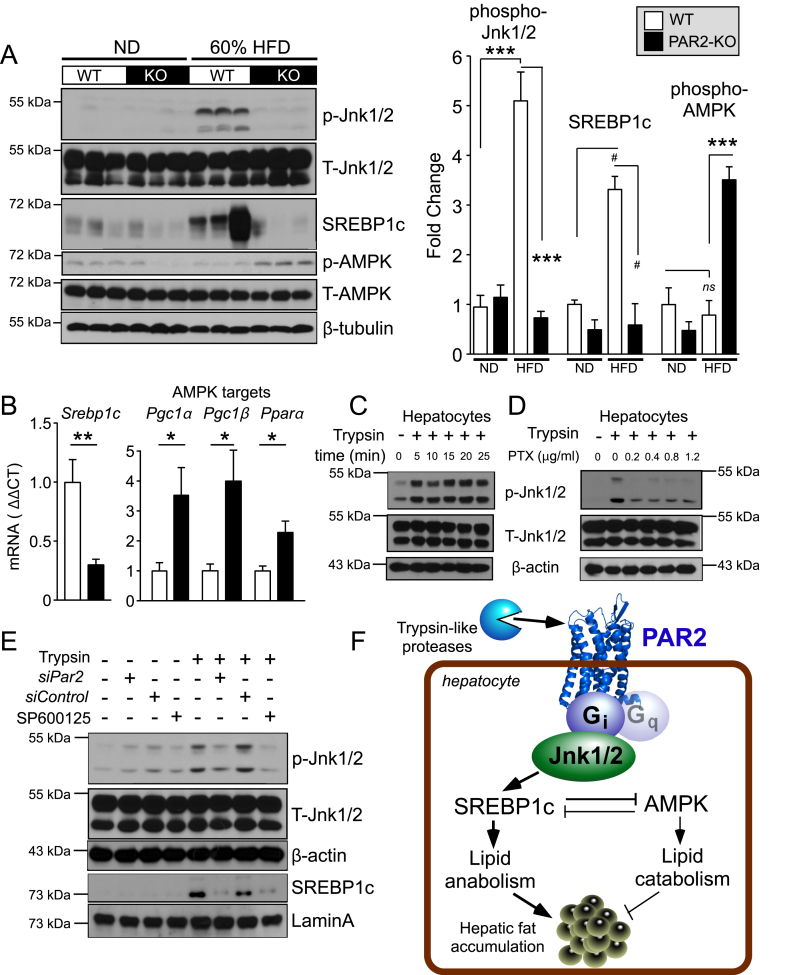
References
-
- Younossi Z.M. Non-alcoholic fatty liver disease - a global public health perspective. Journal of Hepatology. 2018;70(3):531–544. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
