

Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion
- PMID: 31668988
- PMCID: PMC7056924
- DOI: 10.1016/j.molmet.2019.08.021
Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion
Abstract
Background: It has been known for close to a century that, on average, tumors have a metabolism that is different from those found in healthy tissues. Typically, tumors show a biosynthetic metabolism that distinguishes itself by engaging in large scale aerobic glycolysis, heightened flux through the pentose phosphate pathway, and increased glutaminolysis among other means. However, it is becoming equally clear that non tumorous tissues at times can engage in similar metabolism, while tumors show a high degree of metabolic flexibility reacting to cues, and stresses in their local environment.
Scope of the review: In this review, we want to scrutinize historic and recent research on metabolism, comparing and contrasting oncogenic and physiological metabolic states. This will allow us to better define states of bona fide tumor metabolism. We will further contextualize the stress response and the metabolic evolutionary trajectory seen in tumors, and how these contribute to tumor progression. Lastly, we will analyze the implications of these characteristics with respect to therapy response.
Major conclusions: In our review, we argue that there is not one single oncogenic state, but rather a diverse set of oncogenic states. These are grounded on a physiological proliferative/wound healing program but distinguish themselves due to their large scale of proliferation, mutations, and transcriptional changes in key metabolic pathways, and the adaptations to widespread stress signals within tumors. We find evidence for the necessity of metabolic flexibility and stress responses in tumor progression and how these responses in turn shape oncogenic progression. Lastly, we find evidence for the notion that the metabolic adaptability of tumors frequently frustrates therapeutic interventions.
Keywords: Central carbon metabolism; Metabolic flexibility; Tumour metabolism; Tumourigenesis.
Copyright © 2019 The Francis Crick Institute. Published by Elsevier GmbH.. All rights reserved.
Figures
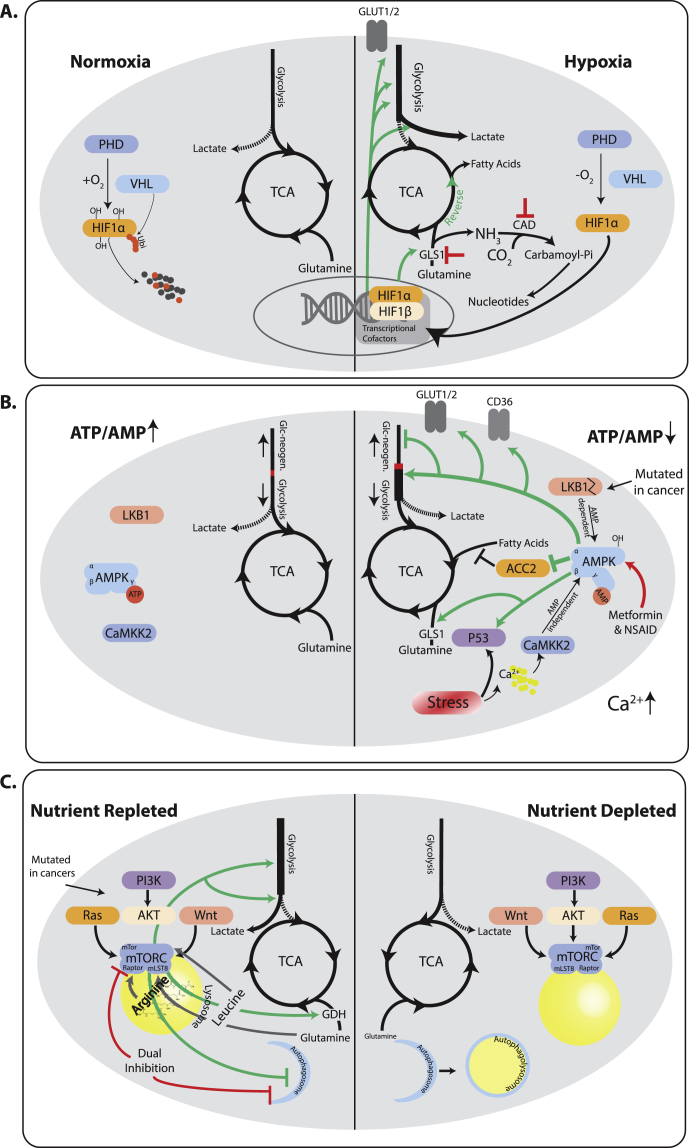
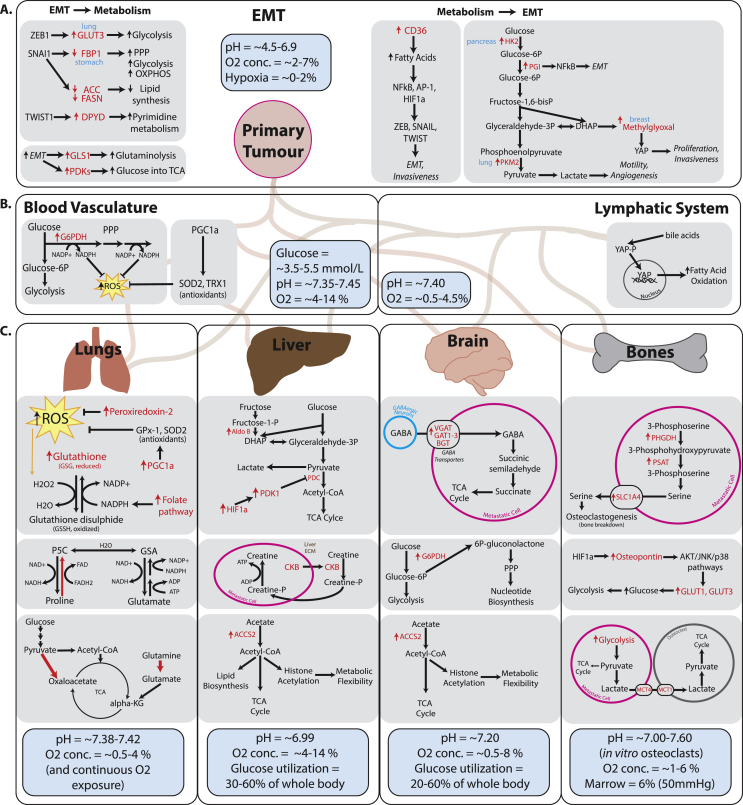
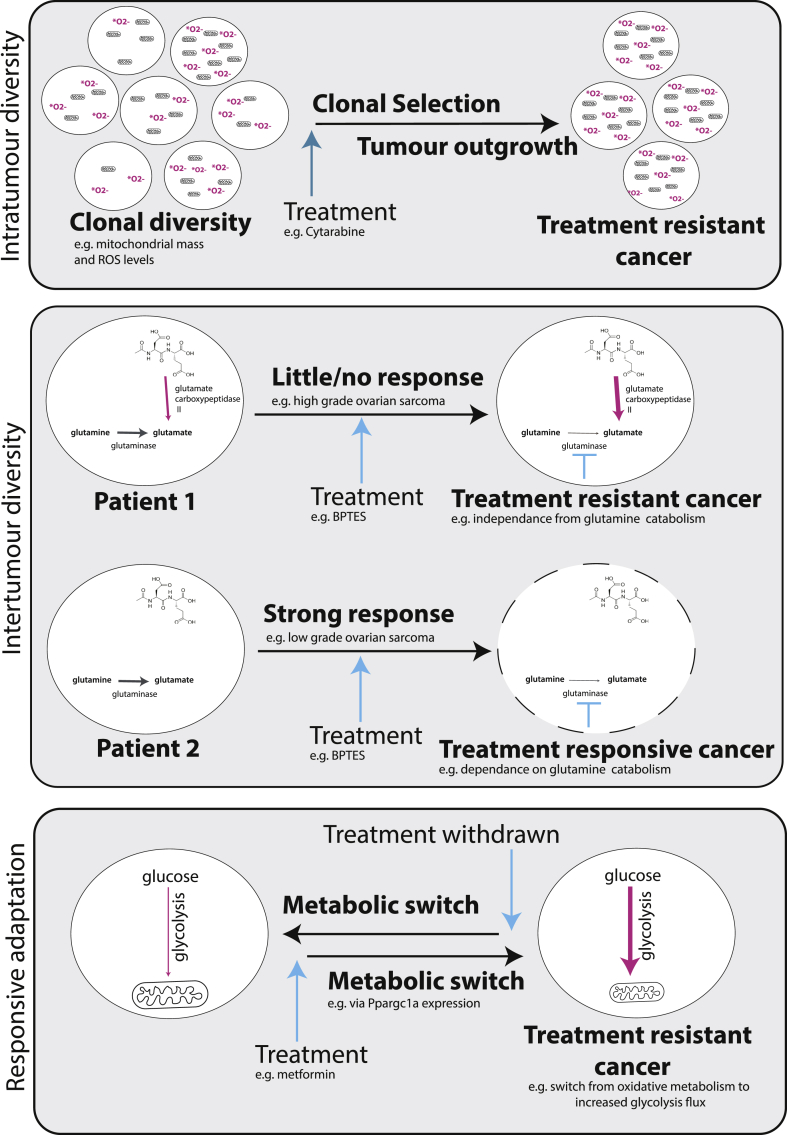
References
-
- Warburg O., Posener K., Negelein E. The metabolism of cancer cells. Biochemische Zeitschrift. 1924;152:319–344.
-
- Gullino P.M., Clark S.H., Grantham F.H. The interstitial fluid of solid tumors. Cancer Research. 1964;24:780–794. - PubMed
-
- Mithieux G. New data and concepts on glutamine and glucose metabolism in the gut. Current Opinion in Clinical Nutrition and Metabolic Care. 2001;4(4):267–271. - PubMed
-
- van Hall G. Lactate kinetics in human tissues at rest and during exercise. Acta Physiologica. 2010;199(4):499–508. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
