

Pathogenesis of SCA3 and implications for other polyglutamine diseases
- PMID: 31669734
- PMCID: PMC6980715
- DOI: 10.1016/j.nbd.2019.104635
Pathogenesis of SCA3 and implications for other polyglutamine diseases
Abstract
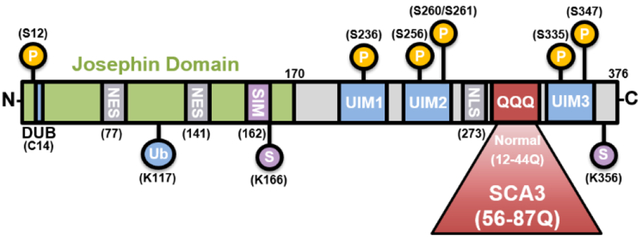
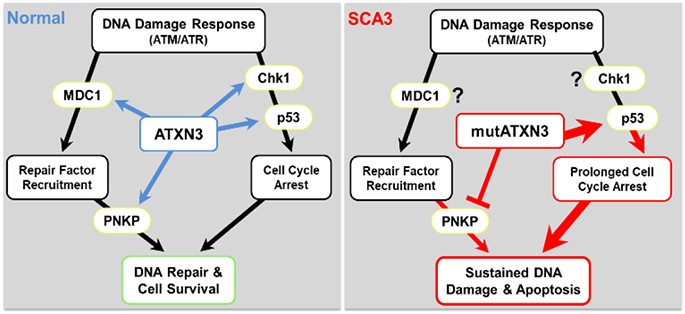
Tandem repeat diseases include the neurodegenerative disorders known as polyglutamine (polyQ) diseases, caused by CAG repeat expansions in the coding regions of the respective disease genes. The nine known polyQ disease include Huntington's disease (HD), dentatorubral-pallidoluysian atrophy (DRPLA), spinal bulbar muscular atrophy (SBMA), and six spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17). The underlying disease mechanism in the polyQ diseases is thought principally to reflect dominant toxic properties of the disease proteins which, when harboring a polyQ expansion, differentially interact with protein partners and are prone to aggregate. Among the polyQ diseases, SCA3 is the most common SCA, and second to HD in prevalence worldwide. Here we summarize current understanding of SCA3 disease mechanisms within the broader context of the broader polyQ disease field. We emphasize properties of the disease protein, ATXN3, and new discoveries regarding three potential pathogenic mechanisms: 1) altered protein homeostasis; 2) DNA damage and dysfunctional DNA repair; and 3) nonneuronal contributions to disease. We conclude with an overview of the therapeutic implications of recent mechanistic insights.
Keywords: ATXN3; Ataxin-3; Deubiquitinase; MJD; Machado Joseph disease; Neurodegenerative disease; Polyglutamine disease; SCA3; Spinocerebellar Ataxia.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Almeida B, Abreu IA, Matos CA, Fraga JS, Fernandes S, Macedo MG, Gutierrez-Gallego R, Pereira PJ, Carvalho AL, and Macedo-Ribeiro S. 2015. 'SUMOylation of the brain-predominant Ataxin-3 isoform modulates its interaction with p97', Biochim Biophys Acta, 1852: 1950–9. - PubMed
-
- Alves S, Nascimento-Ferreira I, Auregan G, Hassig R, Dufour N, Brouillet E, Pedroso de Lima MC, Hantraye P, Pereira de Almeida L, and Deglon N. 2008. 'Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease', PloS one, 3: e3341. - PMC - PubMed
-
- Alves S, Nascimento-Ferreira I, Dufour N, Hassig R, Auregan G, Nobrega C, Brouillet E, Hantraye P, Pedroso de Lima MC, Deglon N, and de Almeida LP. 2010. 'Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3?', Hum Mol Genet, 19: 2380–94. - PubMed
-
- Amirifar P, Ranjouri MR, Yazdani R, Abolhassani H, and Aghamohammadi A. 2019. 'Ataxia-telangiectasia: A review of clinical features and molecular pathology', Pediatr Allergy Immunol, 30: 277–88. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
