

Tumor protein D54 defines a new class of intracellular transport vesicles
- PMID: 31672706
- PMCID: PMC7039206
- DOI: 10.1083/jcb.201812044
Tumor protein D54 defines a new class of intracellular transport vesicles
Abstract
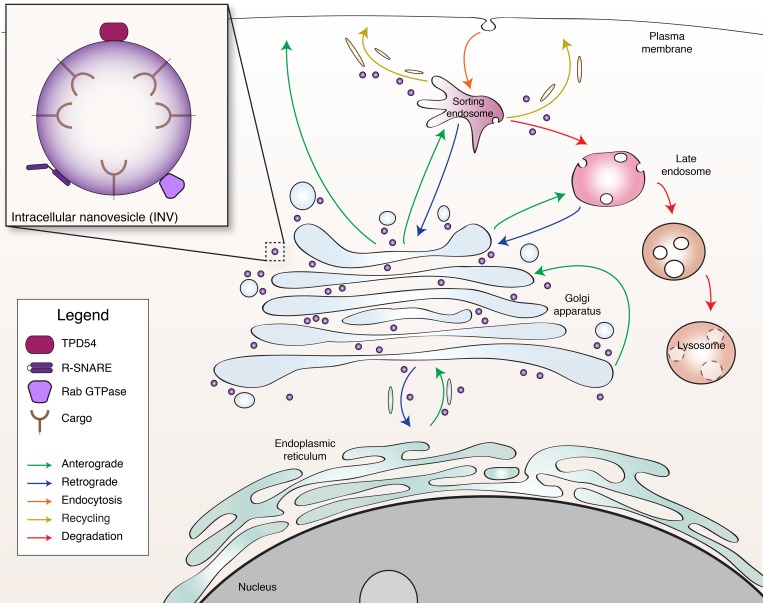
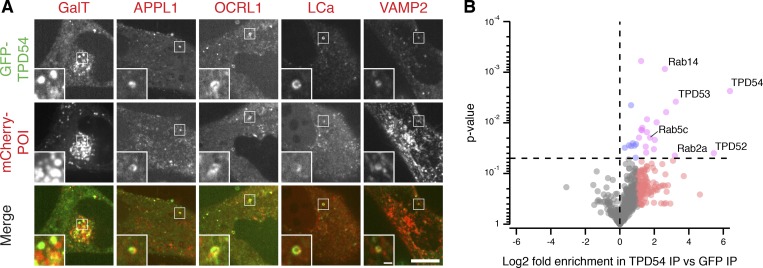
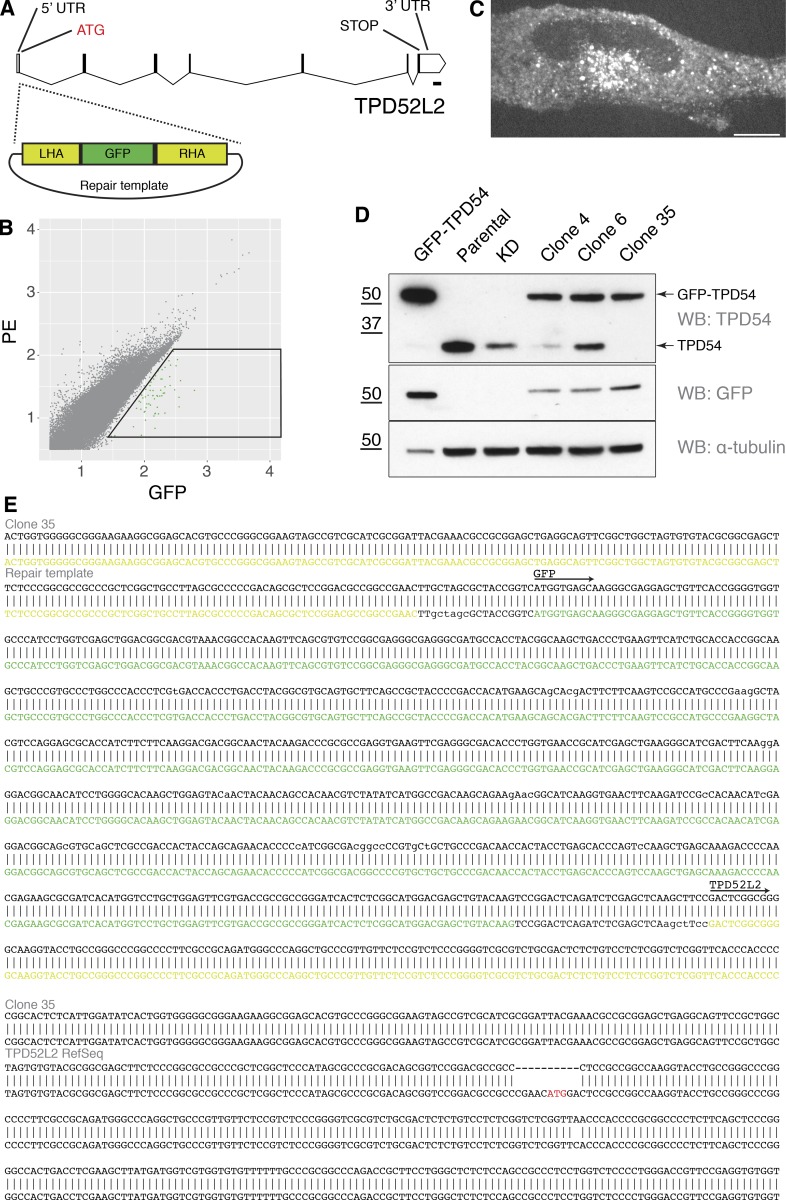
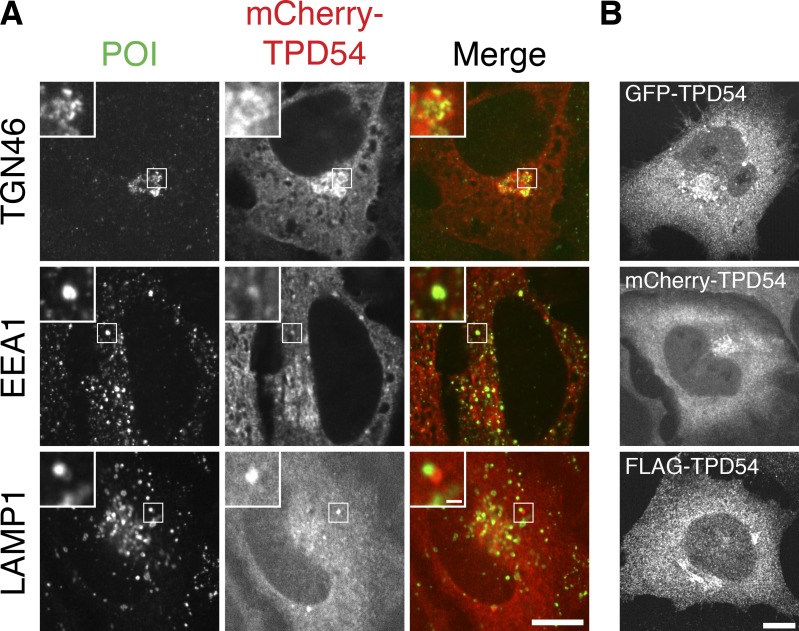
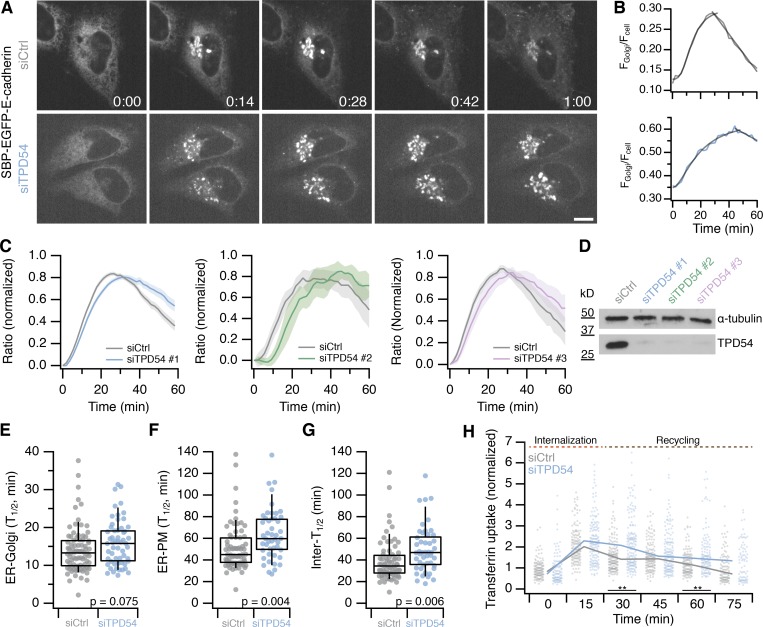
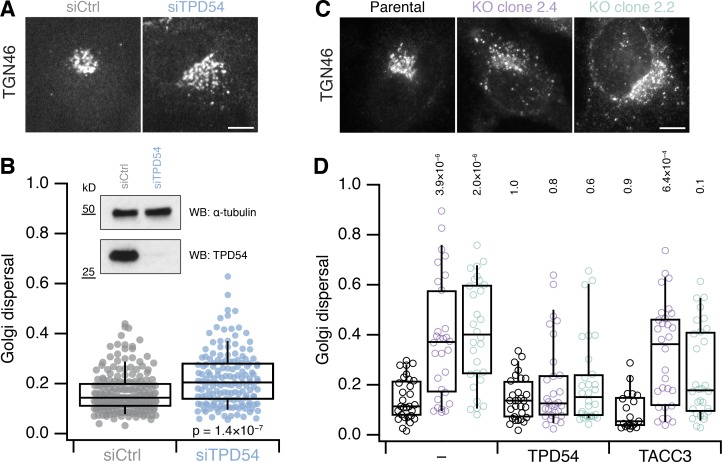
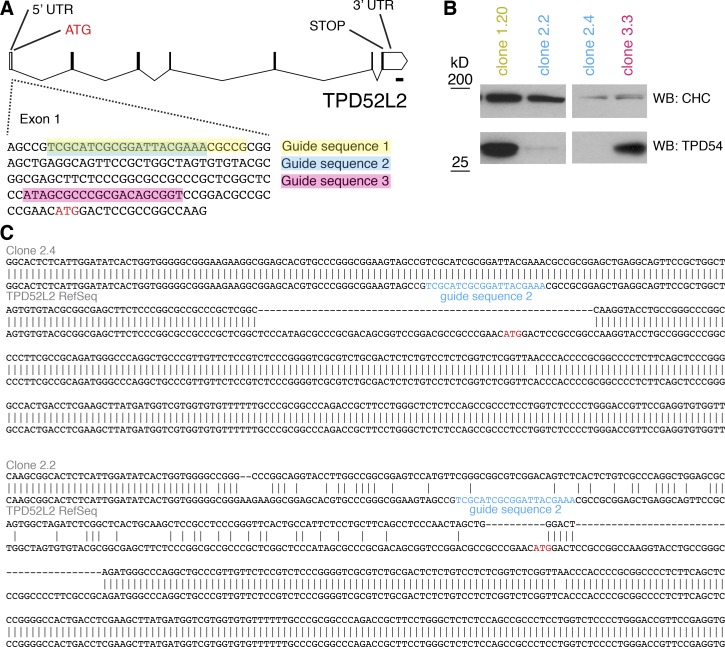
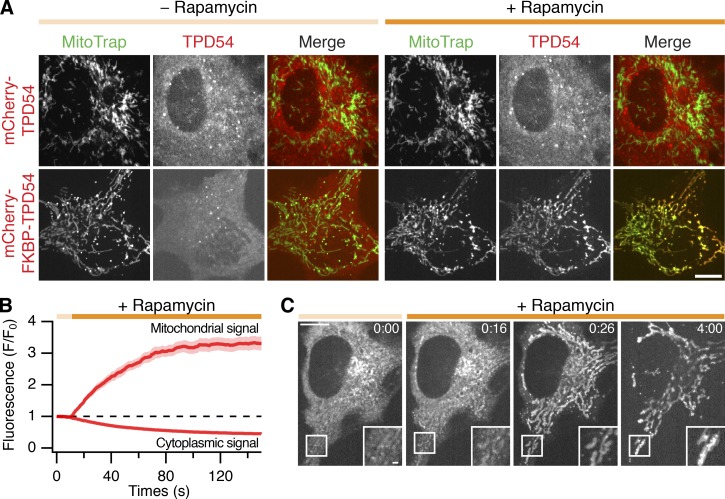
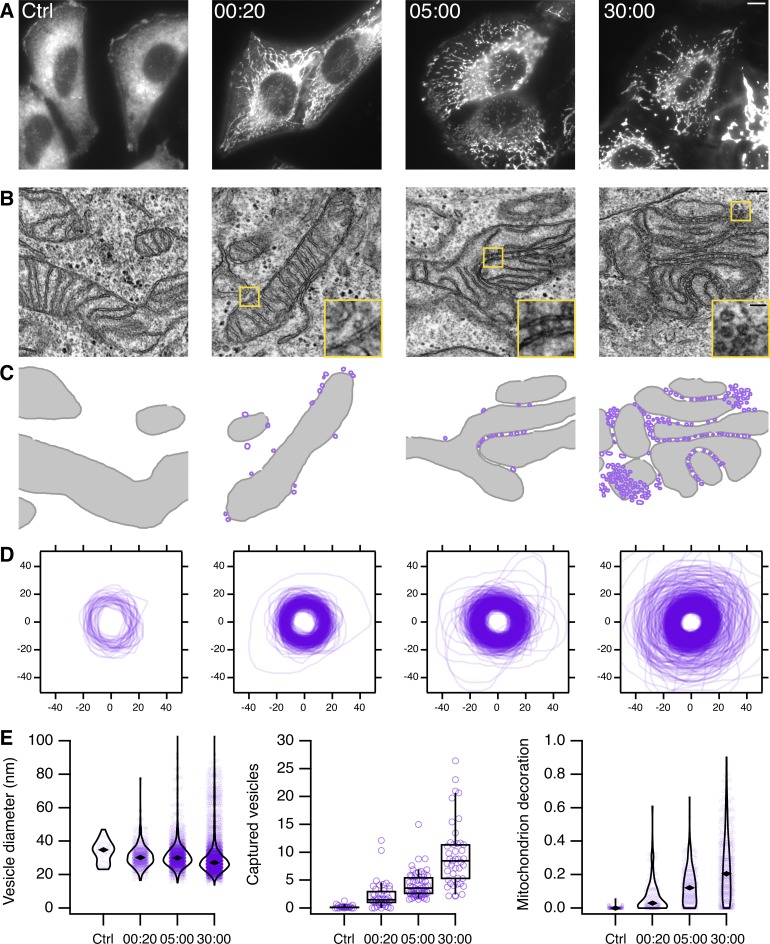
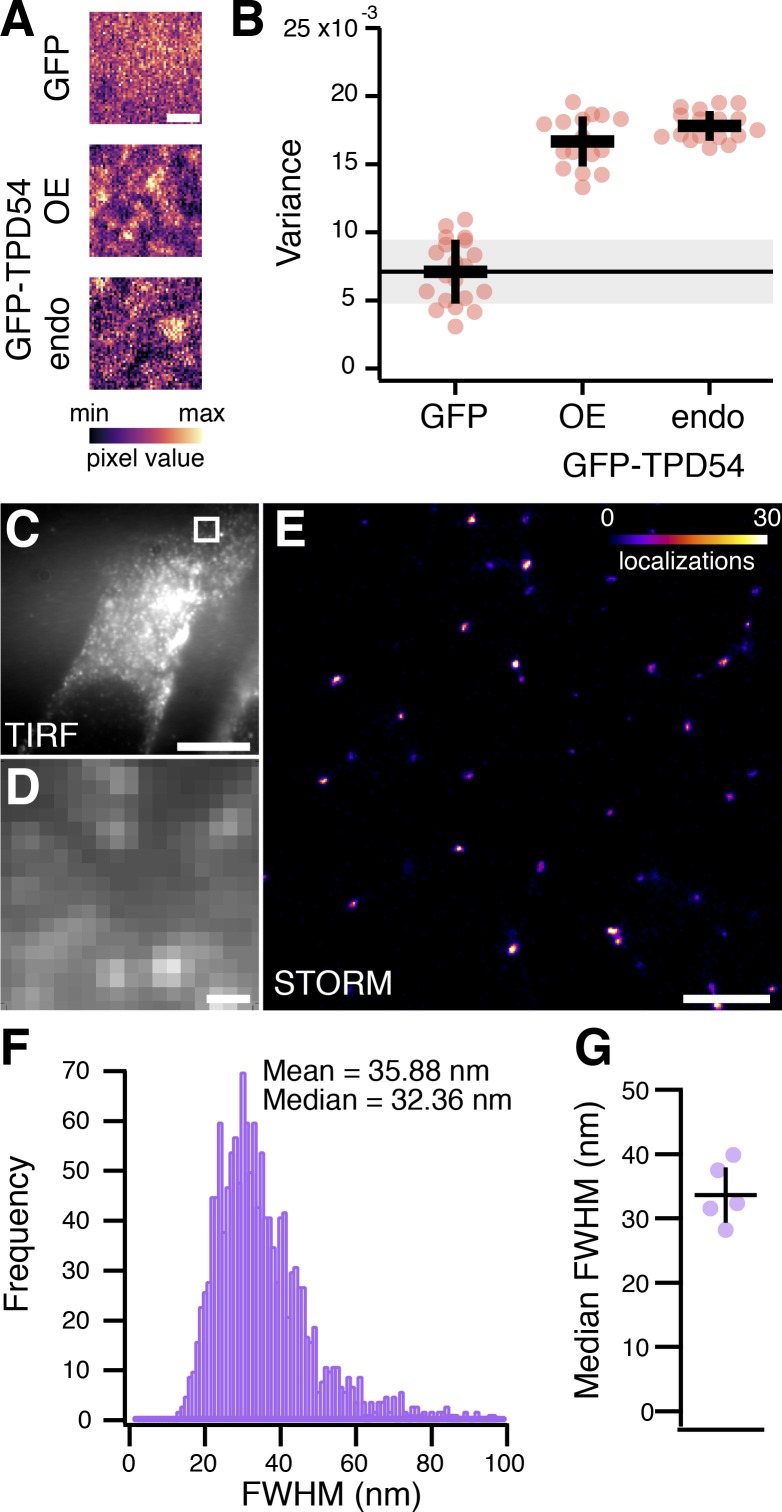
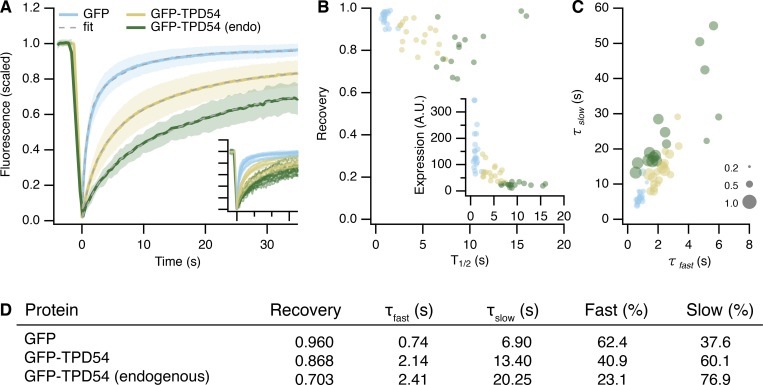
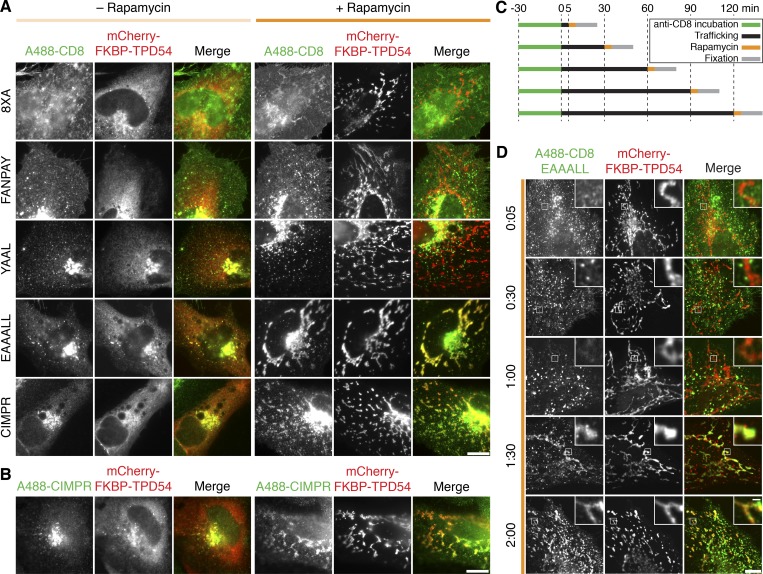
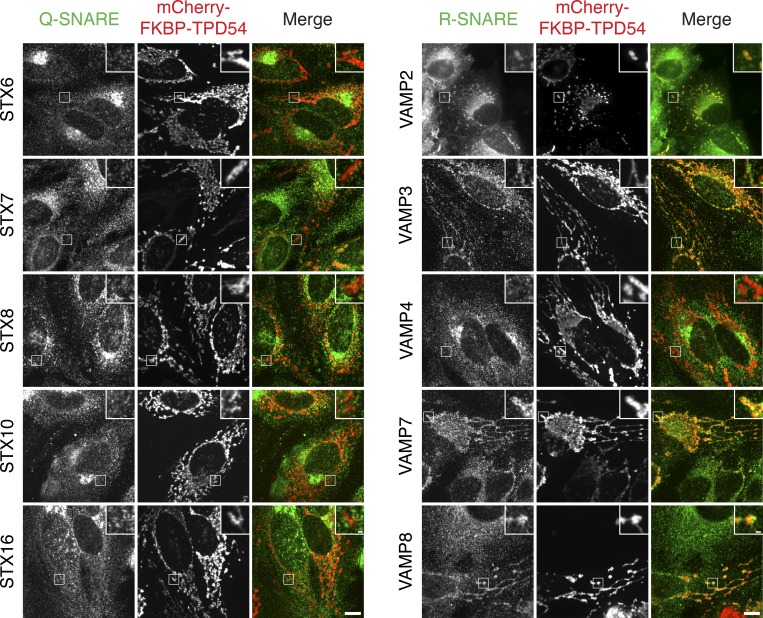
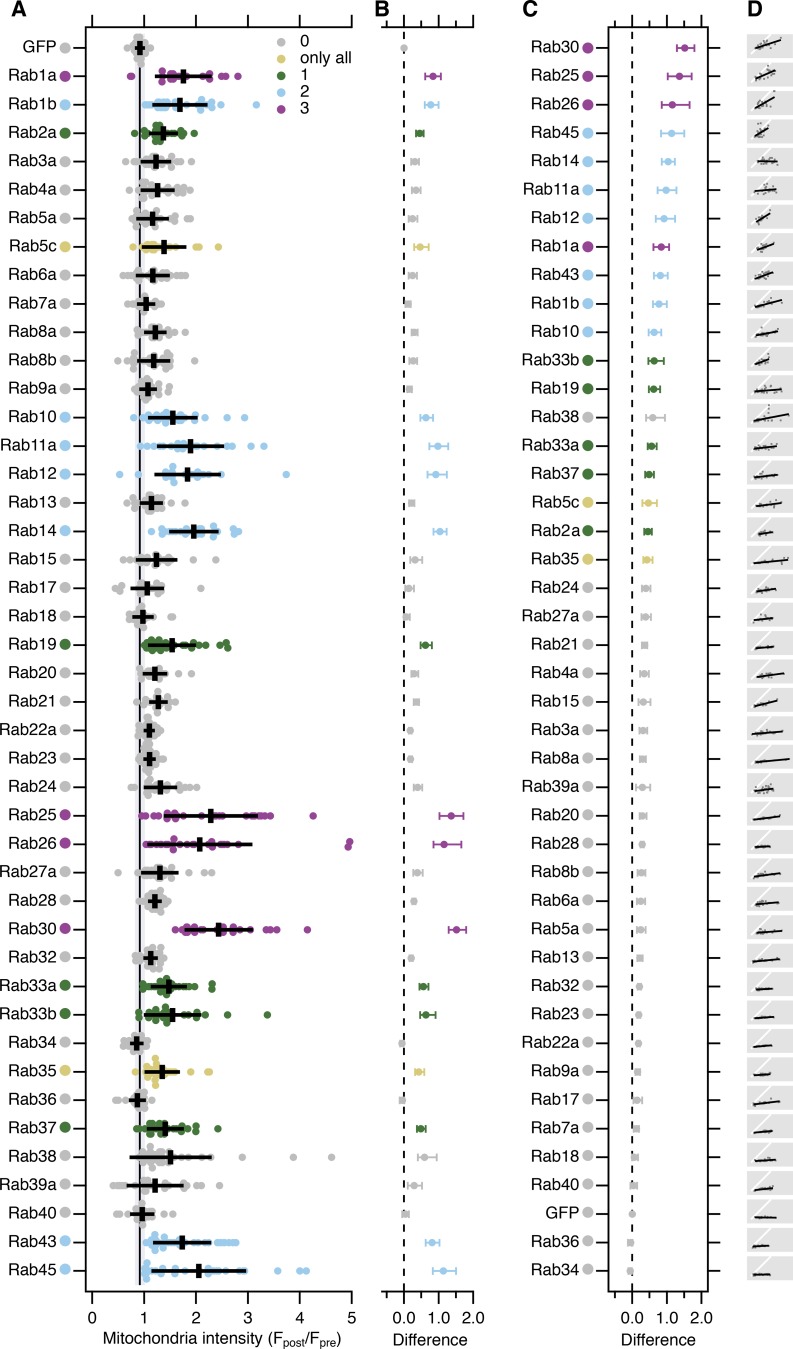
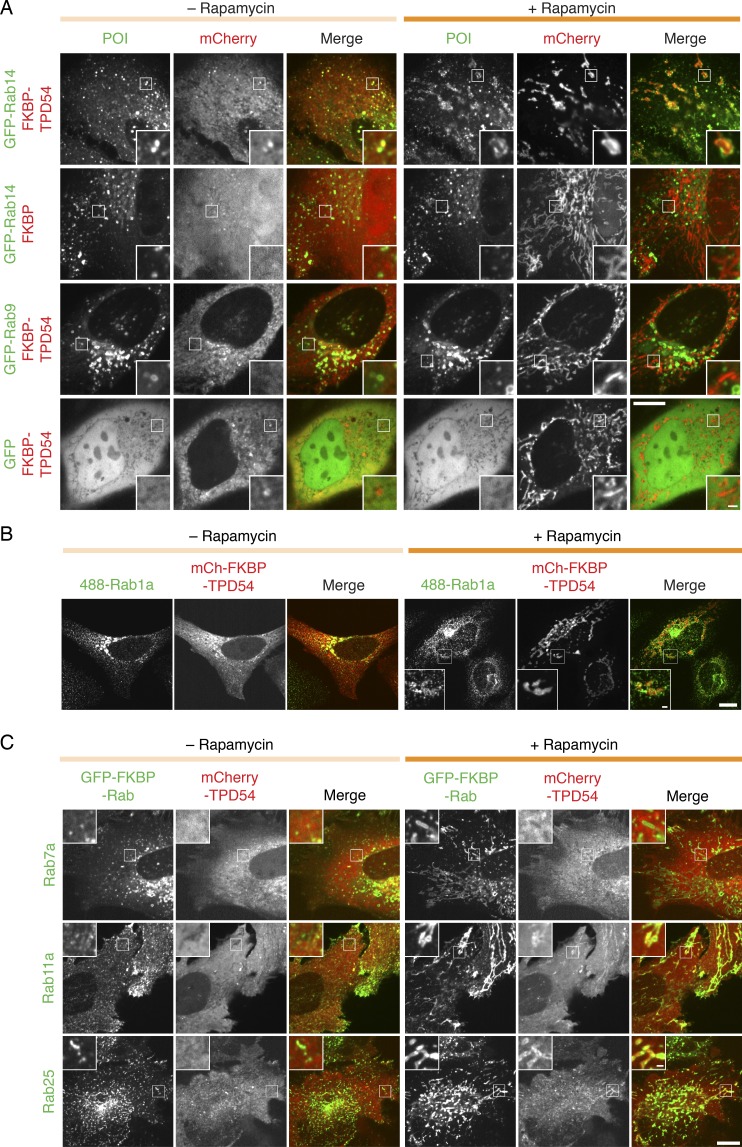
Transport of proteins and lipids from one membrane compartment to another is via intracellular vesicles. We investigated the function of tumor protein D54 (TPD54/TPD52L2) and found that TPD54 was involved in multiple membrane trafficking pathways: anterograde traffic, recycling, and Golgi integrity. To understand how TPD54 controls these diverse functions, we used an inducible method to reroute TPD54 to mitochondria. Surprisingly, this manipulation resulted in the capture of many small vesicles (30 nm diameter) at the mitochondrial surface. Super-resolution imaging confirmed the presence of similarly sized TPD54-positive structures under normal conditions. It appears that TPD54 defines a new class of transport vesicle, which we term intracellular nanovesicles (INVs). INVs meet three criteria for functionality. They contain specific cargo, they have certain R-SNAREs for fusion, and they are endowed with a variety of Rab GTPases (16 out of 43 tested). The molecular heterogeneity of INVs and the diverse functions of TPD54 suggest that INVs have various membrane origins and a number of destinations. We propose that INVs are a generic class of transport vesicle that transfer cargo between these varied locations.
© 2019 Larocque et al.
Figures

References
-
- Biesemann C., Grønborg M., Luquet E., Wichert S.P., Bernard V., Bungers S.R., Cooper B., Varoqueaux F., Li L., Byrne J.A., et al. . 2014. Proteomic screening of glutamatergic mouse brain synaptosomes isolated by fluorescence activated sorting. EMBO J. 33:157–170. 10.1002/embj.201386120 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
