

The Signaling Pathways Project, an integrated 'omics knowledgebase for mammalian cellular signaling pathways
- PMID: 31672983
- PMCID: PMC6823428
- DOI: 10.1038/s41597-019-0193-4
The Signaling Pathways Project, an integrated 'omics knowledgebase for mammalian cellular signaling pathways
Abstract
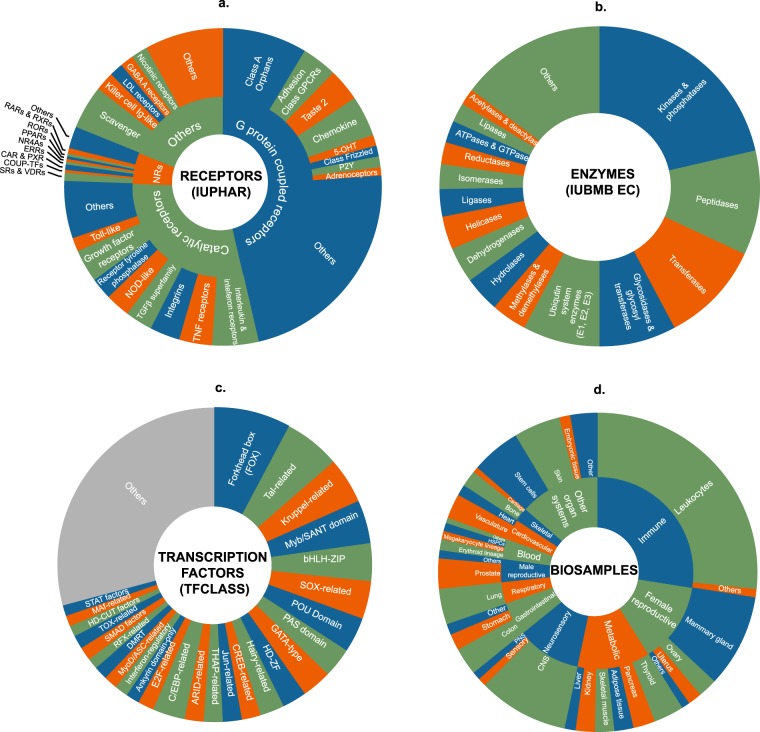
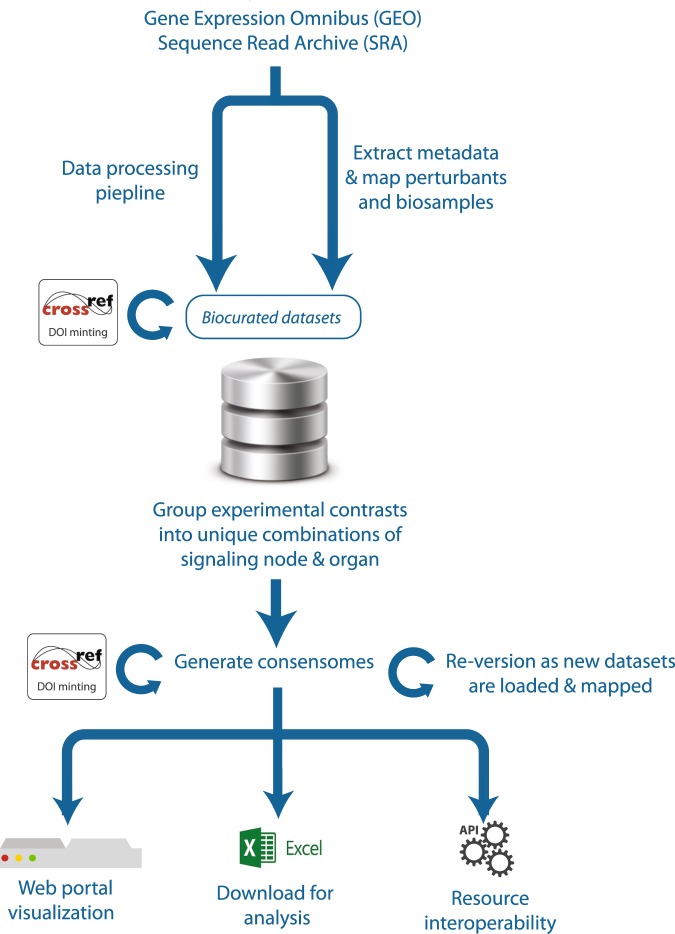
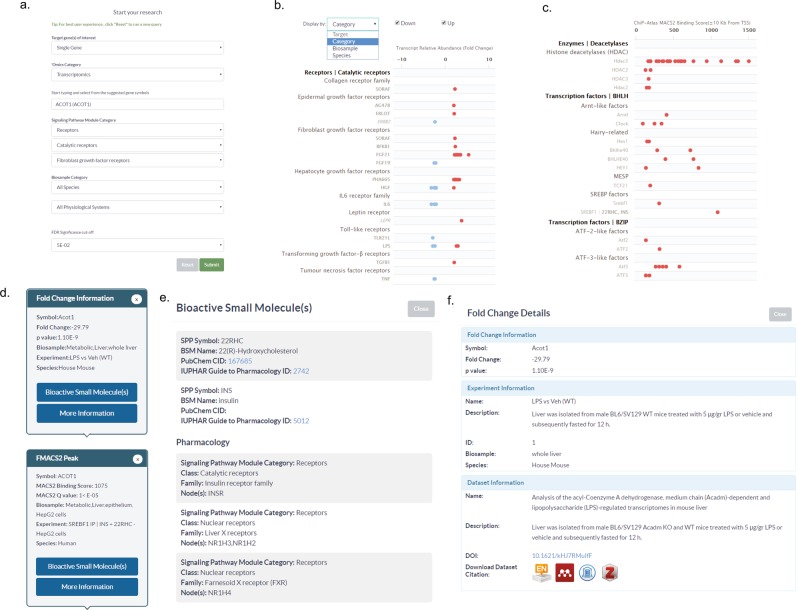
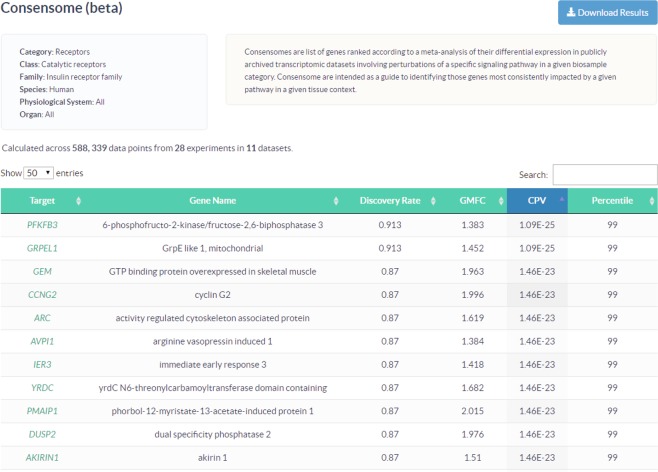
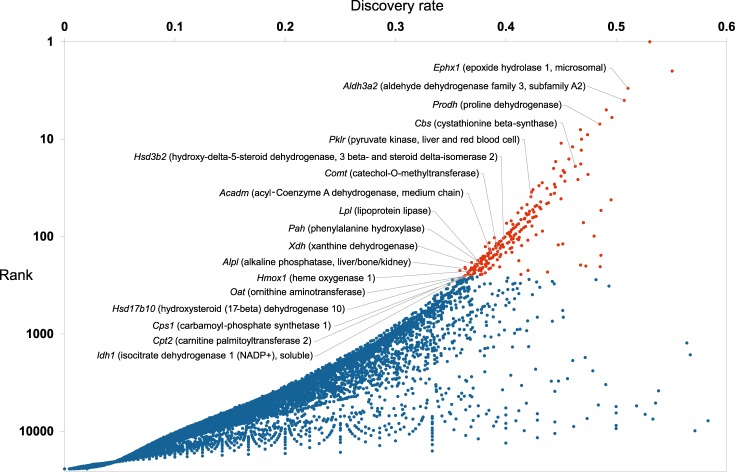
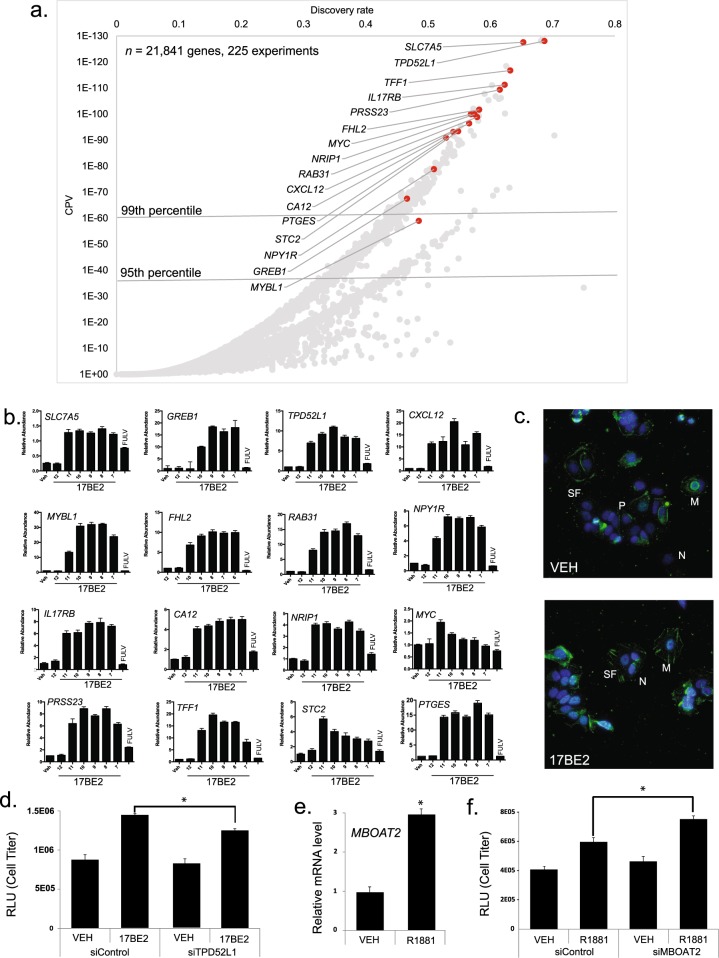
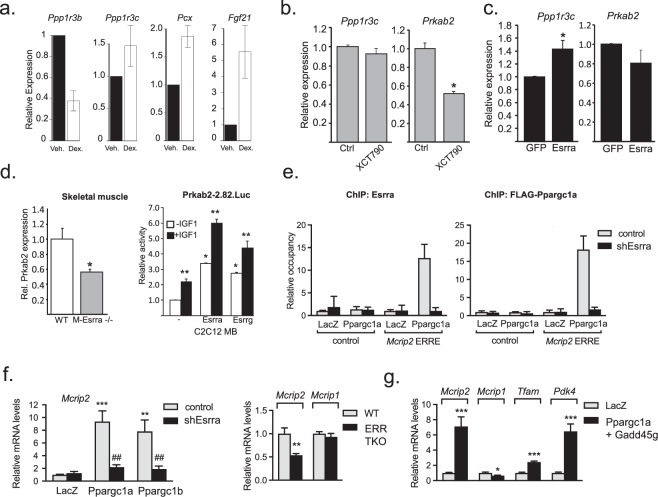
Mining of integrated public transcriptomic and ChIP-Seq (cistromic) datasets can illuminate functions of mammalian cellular signaling pathways not yet explored in the research literature. Here, we designed a web knowledgebase, the Signaling Pathways Project (SPP), which incorporates community classifications of signaling pathway nodes (receptors, enzymes, transcription factors and co-nodes) and their cognate bioactive small molecules. We then mapped over 10,000 public transcriptomic or cistromic experiments to their pathway node or biosample of study. To enable prediction of pathway node-gene target transcriptional regulatory relationships through SPP, we generated consensus 'omics signatures, or consensomes, which ranked genes based on measures of their significant differential expression or promoter occupancy across transcriptomic or cistromic experiments mapped to a specific node family. Consensomes were validated using alignment with canonical literature knowledge, gene target-level integration of transcriptomic and cistromic data points, and in bench experiments confirming previously uncharacterized node-gene target regulatory relationships. To expose the SPP knowledgebase to researchers, a web browser interface was designed that accommodates numerous routine data mining strategies. SPP is freely accessible at https://www.signalingpathways.org .
Conflict of interest statement
Charles Foulds has equity in Coactigon, Inc.
Figures
References
-
- Becnel Lauren B., Ochsner Scott A., Darlington Yolanda F., McOwiti Apollo, Kankanamge Wasula H., Dehart Michael, Naumov Alexey, McKenna Neil J. Discovering relationships between nuclear receptor signaling pathways, genes, and tissues in Transcriptomine. Science Signaling. 2017;10(476):eaah6275. doi: 10.1126/scisignal.aah6275. - DOI - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
