

NAD+ homeostasis in renal health and disease
- PMID: 31673160
- PMCID: PMC7223841
- DOI: 10.1038/s41581-019-0216-6
NAD+ homeostasis in renal health and disease
Abstract
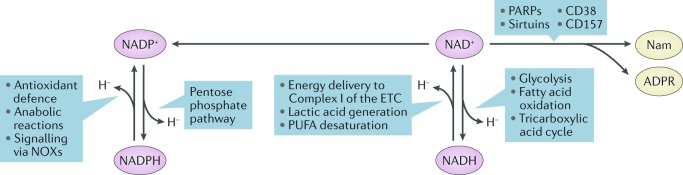
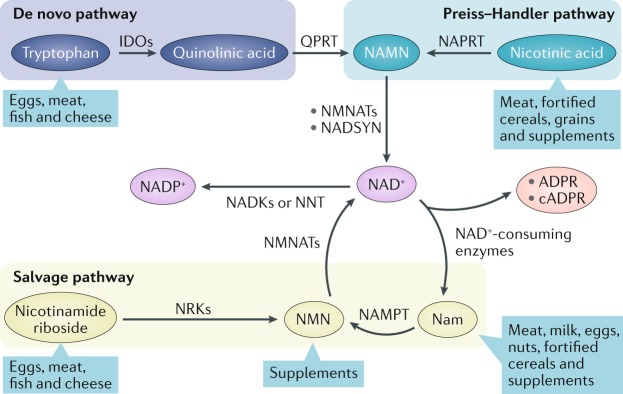
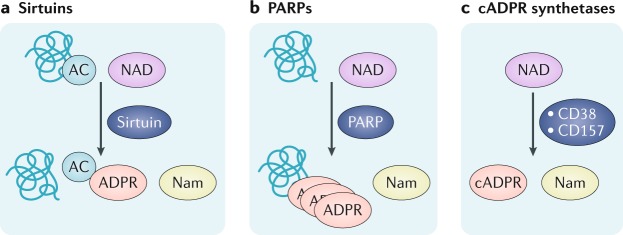
The mammalian kidney relies on abundant mitochondria in the renal tubule to generate sufficient ATP to provide the energy required for constant reclamation of solutes from crude blood filtrate. The highly metabolically active cells of the renal tubule also pair their energetic needs to the regulation of diverse cellular processes, including energy generation, antioxidant responses, autophagy and mitochondrial quality control. Nicotinamide adenine dinucleotide (NAD+) is essential not only for the harvesting of energy from substrates but also for an array of regulatory reactions that determine cellular health. In acute kidney injury (AKI), substantial decreases in the levels of NAD+ impair energy generation and, ultimately, the core kidney function of selective solute transport. Conversely, augmentation of NAD+ may protect the kidney tubule against diverse acute stressors. For example, NAD+ augmentation can ameliorate experimental AKI triggered by ischaemia-reperfusion, toxic injury and systemic inflammation. NAD+-dependent maintenance of renal tubular metabolic health may also attenuate long-term profibrotic responses that could lead to chronic kidney disease. Further understanding of the genetic, environmental and nutritional factors that influence NAD+ biosynthesis and renal resilience may lead to novel approaches for the prevention and treatment of kidney disease.
Conflict of interest statement
S.M.P. is an inventor of patent filings from Beth Israel Deaconess Medical Center. He holds equity in Raksana, serves on the Scientific Advisory Board of Aerpio and has received consulting fees from Cytokinetics, Astellas, Janssen, Mission Therapeutics and Aerpio. K.M.R. and E.P.R. declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
