

Uganda Genome Resource Enables Insights into Population History and Genomic Discovery in Africa
- PMID: 31675503
- PMCID: PMC7202134
- DOI: 10.1016/j.cell.2019.10.004
Uganda Genome Resource Enables Insights into Population History and Genomic Discovery in Africa
Abstract
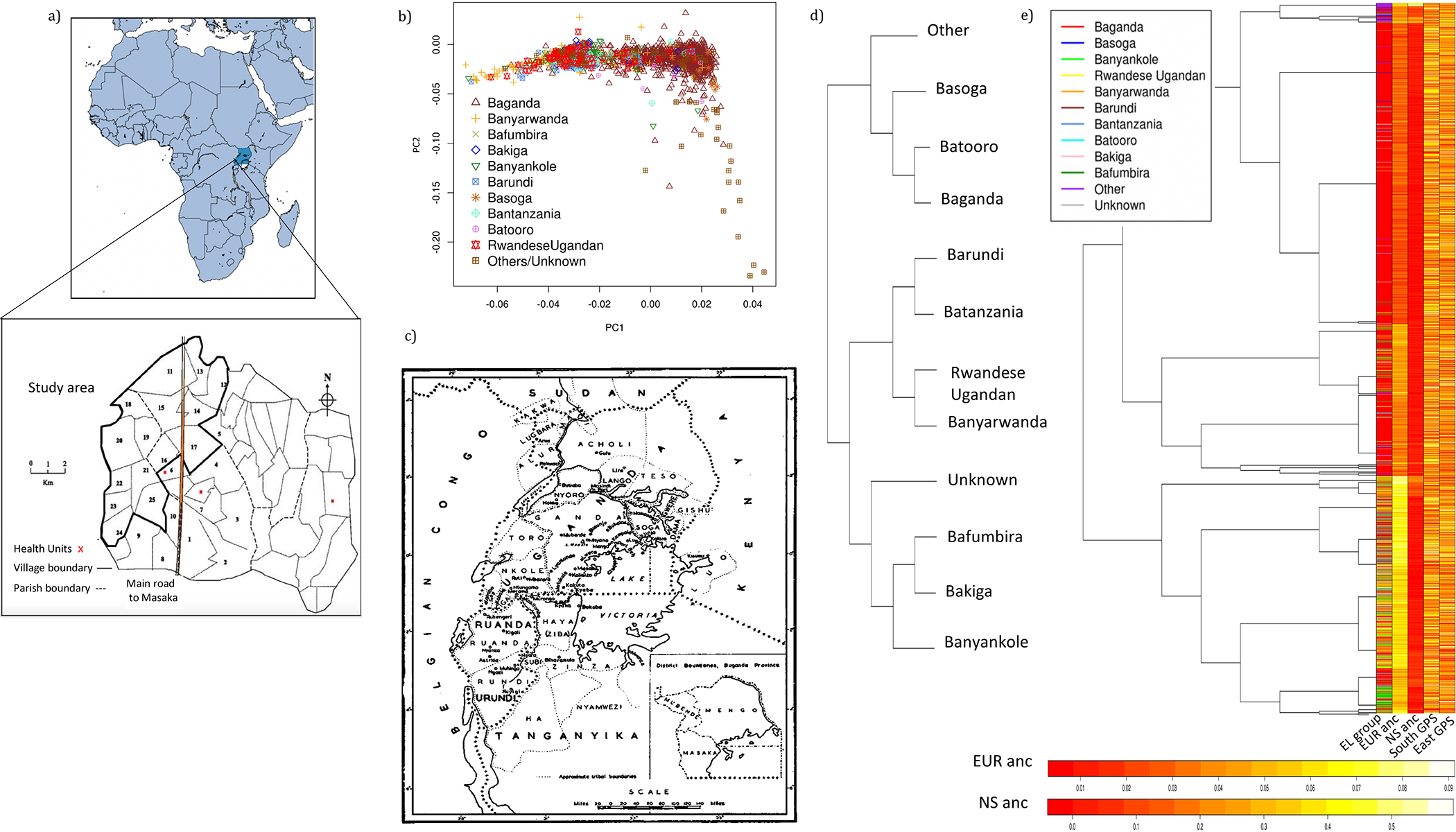
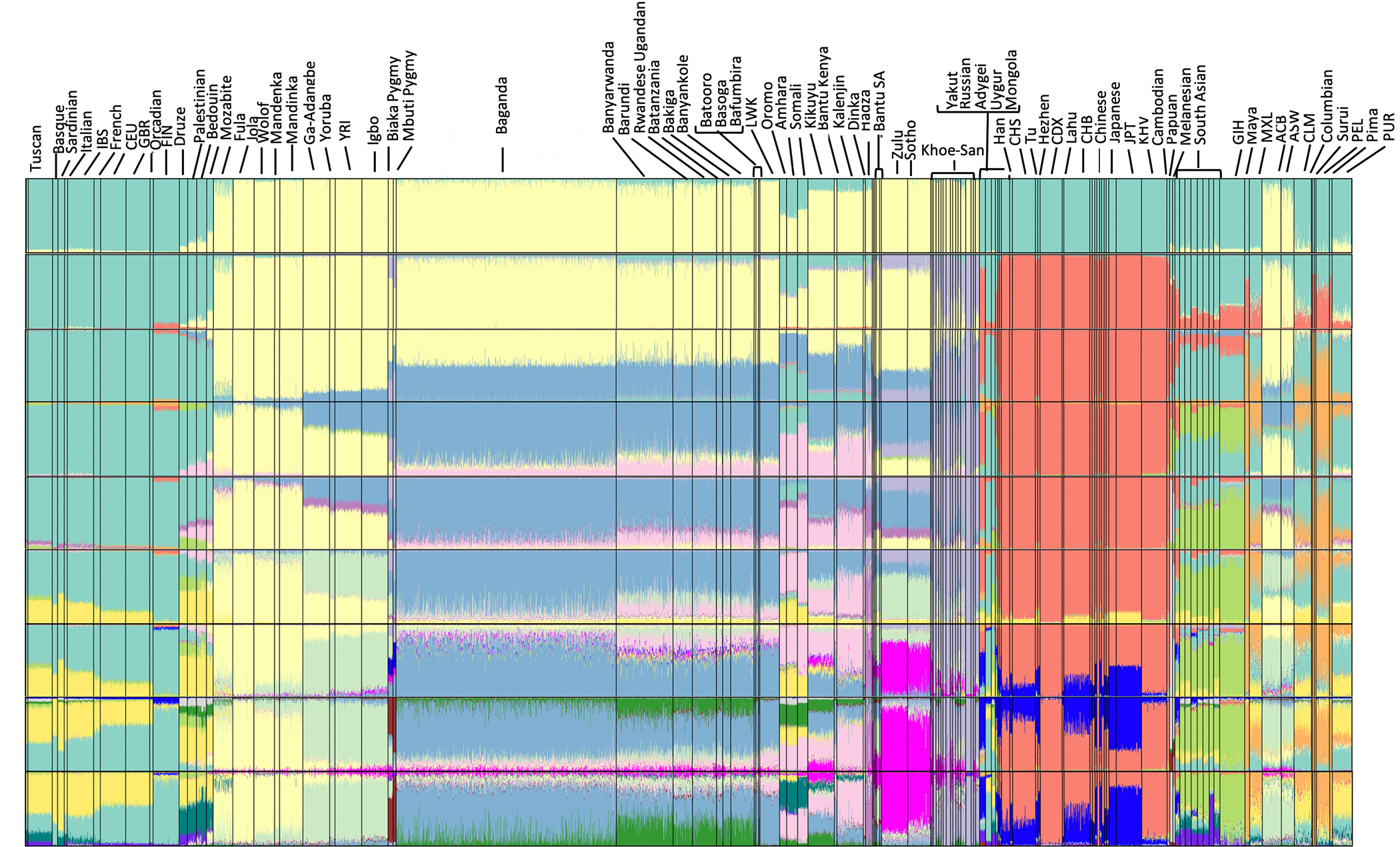
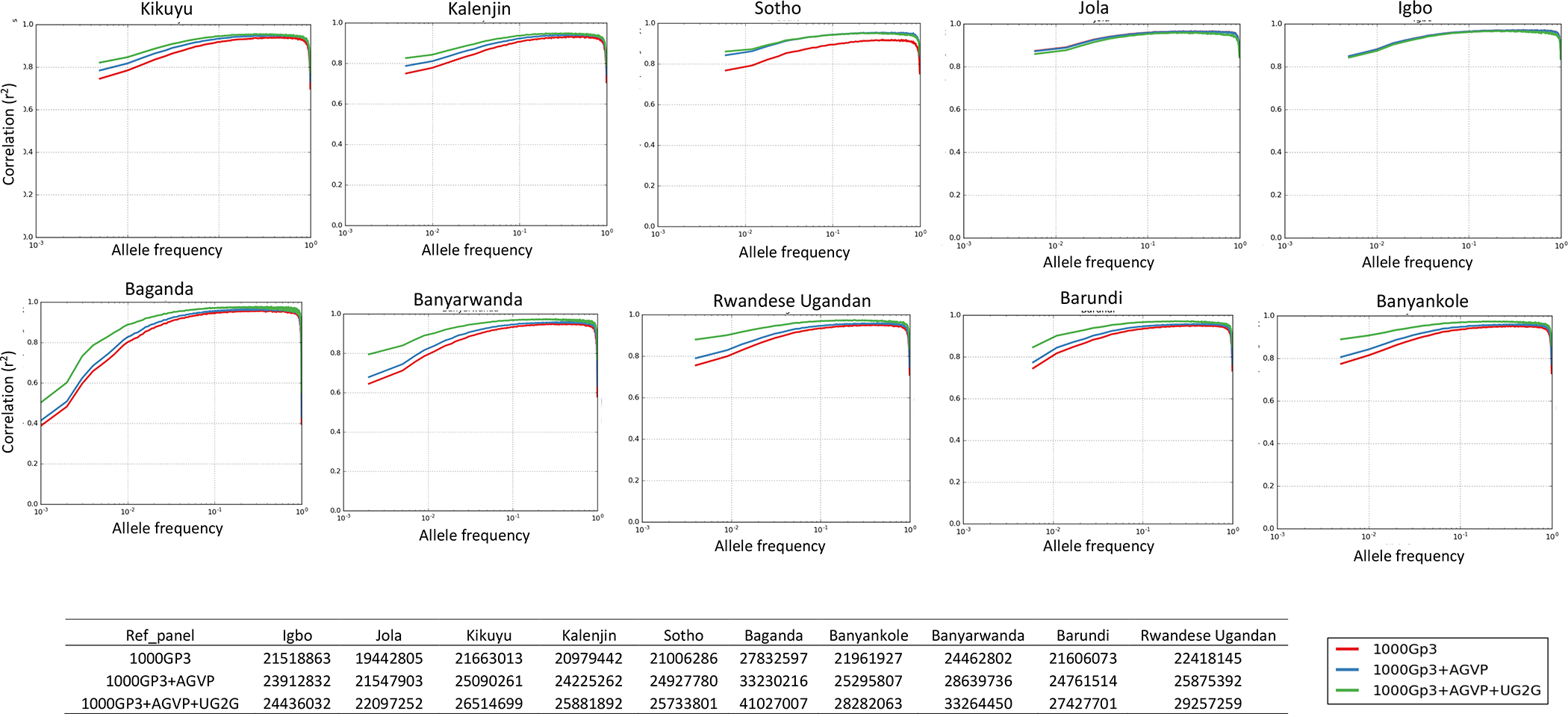
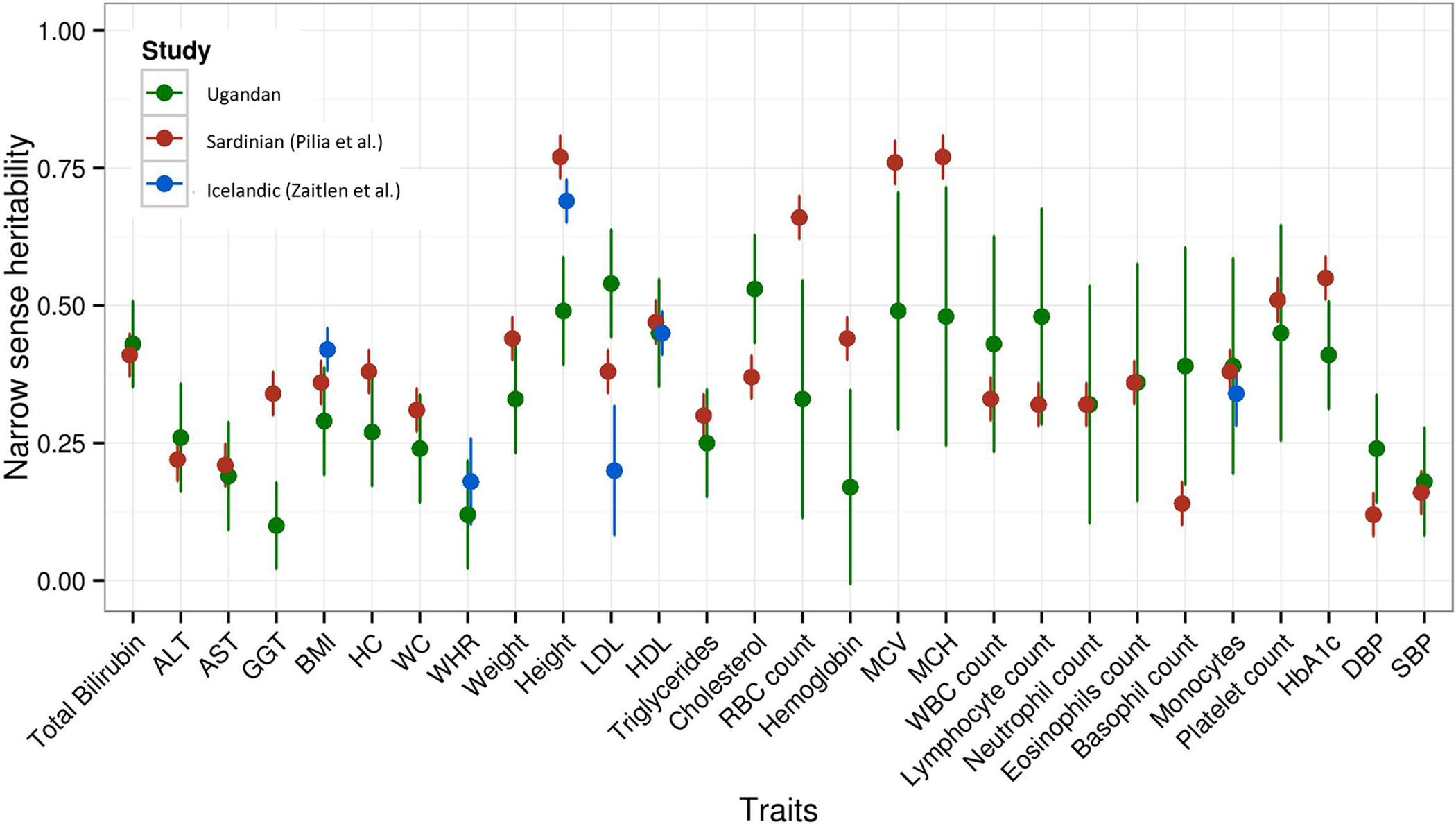
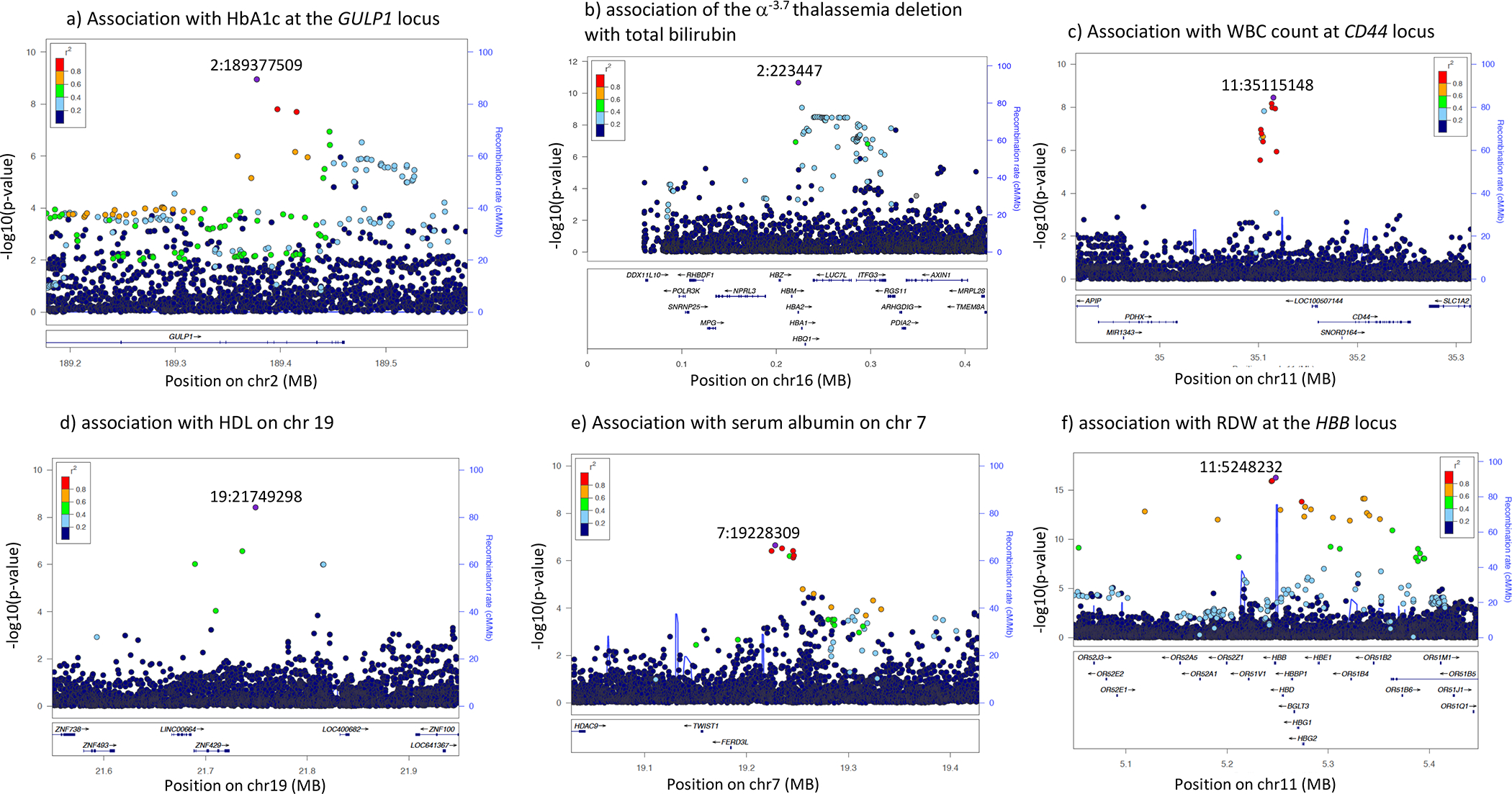
Genomic studies in African populations provide unique opportunities to understand disease etiology, human diversity, and population history. In the largest study of its kind, comprising genome-wide data from 6,400 individuals and whole-genome sequences from 1,978 individuals from rural Uganda, we find evidence of geographically correlated fine-scale population substructure. Historically, the ancestry of modern Ugandans was best represented by a mixture of ancient East African pastoralists. We demonstrate the value of the largest sequence panel from Africa to date as an imputation resource. Examining 34 cardiometabolic traits, we show systematic differences in trait heritability between European and African populations, probably reflecting the differential impact of genes and environment. In a multi-trait pan-African GWAS of up to 14,126 individuals, we identify novel loci associated with anthropometric, hematological, lipid, and glycemic traits. We find that several functionally important signals are driven by Africa-specific variants, highlighting the value of studying diverse populations across the region.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures

Comment in
-
Insights from Ugandan genomes.Nat Rev Genet. 2020 Jan;21(1):4. doi: 10.1038/s41576-019-0194-3. Nat Rev Genet. 2020. PMID: 31695142 No abstract available.
References
-
- Abadie JM, and Koelsch AA (2008). Performance of the Roche second generation hemoglobin A1c immunoassay in the presence of HB-S or HB-C traits. Ann. Clin. Lab. Sci 38, 31–36. - PubMed
-
- Abecasis GR, Cherny SS, Cookson WO, and Cardon LR (2002). Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet 30, 97–101. - PubMed
Publication types
MeSH terms
Grants and funding
- 107743/WT_/Wellcome Trust/United Kingdom
- R21 HL140385/HL/NHLBI NIH HHS/United States
- MC_UU_00027/1/MRC_/Medical Research Council/United Kingdom
- U41 HG006941/HG/NHGRI NIH HHS/United States
- MC_EX_MR/L016273/1/MRC_/Medical Research Council/United Kingdom
- MR/S003711/2/MRC_/Medical Research Council/United Kingdom
- R21 HL123677/HL/NHLBI NIH HHS/United States
- MR/K013491/1/MRC_/Medical Research Council/United Kingdom
- R56 DK104806/DK/NIDDK NIH HHS/United States
- R01 DK117445/DK/NIDDK NIH HHS/United States
- G0901213/MRC_/Medical Research Council/United Kingdom
- MR/S003711/1/MRC_/Medical Research Council/United Kingdom
- G0801566/MRC_/Medical Research Council/United Kingdom
- R01 MD012765/MD/NIMHD NIH HHS/United States
- U01 MH115485/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
