

The impact of short tandem repeat variation on gene expression
- PMID: 31676866
- PMCID: PMC6917484
- DOI: 10.1038/s41588-019-0521-9
The impact of short tandem repeat variation on gene expression
Abstract
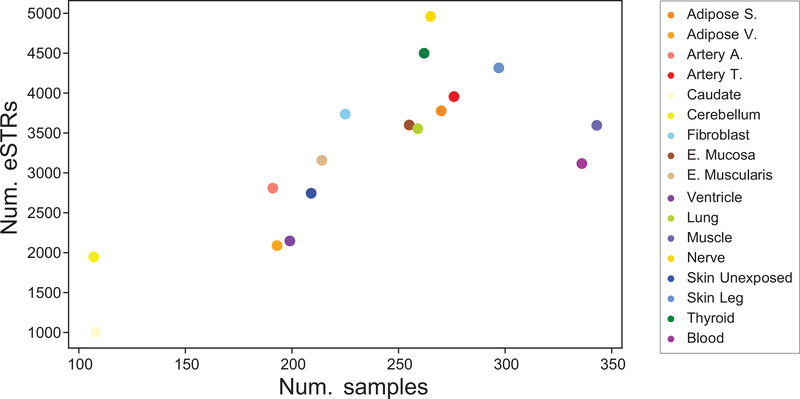
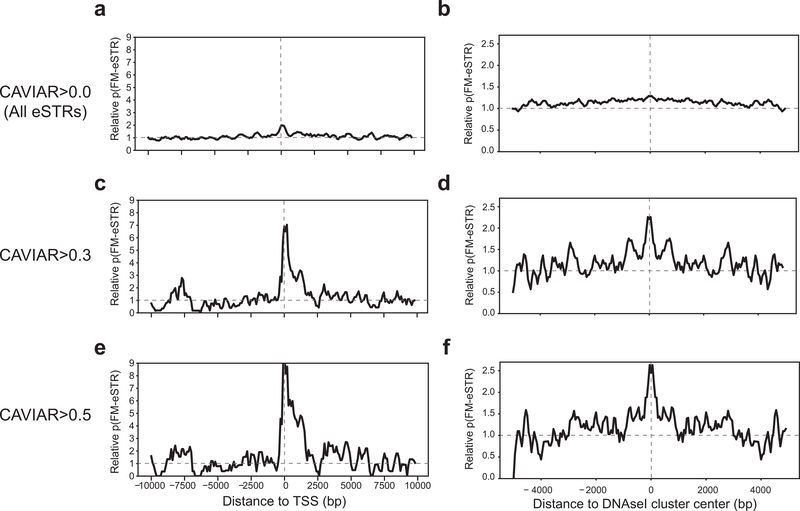
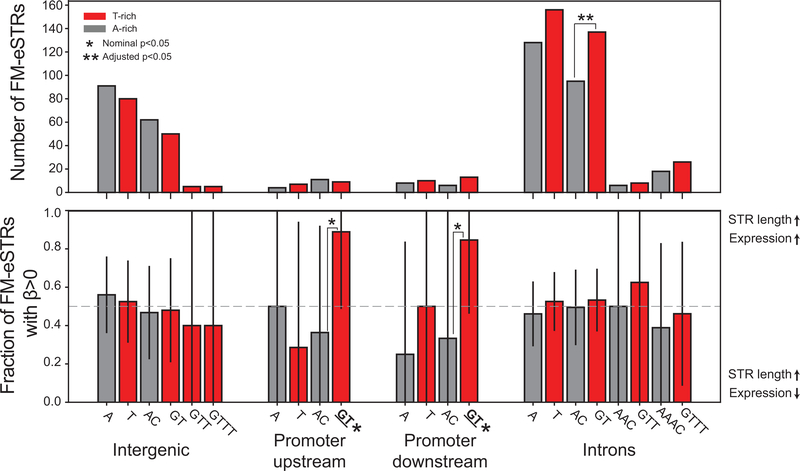
Short tandem repeats (STRs) have been implicated in a variety of complex traits in humans. However, genome-wide studies of the effects of STRs on gene expression thus far have had limited power to detect associations and provide insights into putative mechanisms. Here, we leverage whole-genome sequencing and expression data for 17 tissues from the Genotype-Tissue Expression Project to identify more than 28,000 STRs for which repeat number is associated with expression of nearby genes (eSTRs). We use fine-mapping to quantify the probability that each eSTR is causal and characterize the top 1,400 fine-mapped eSTRs. We identify hundreds of eSTRs linked with published genome-wide association study signals and implicate specific eSTRs in complex traits, including height, schizophrenia, inflammatory bowel disease and intelligence. Overall, our results support the hypothesis that eSTRs contribute to a range of human phenotypes, and our data should serve as a valuable resource for future studies of complex traits.
Conflict of interest statement
Competing Interests
The authors have no competing interests to declare.
Figures











References
References for Main Text
Methods-only references
-
- Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–9 (2006). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
