

Growth failure and treatment in cystic fibrosis
- PMID: 31679733
- PMCID: PMC6934044
- DOI: 10.1016/j.jcf.2019.08.010
Growth failure and treatment in cystic fibrosis
Abstract
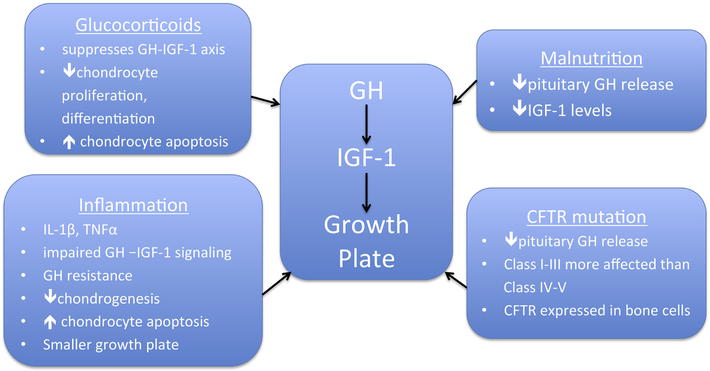
Poor growth has long been a characteristic feature of cystic fibrosis (CF) and is significantly linked to lung function and overall health status. Improvements in pulmonary and nutrition care for patients with cystic fibrosis (CF) have resulted in better growth outcomes; however, height gains have not paralleled the improvements in weight in children with CF, and patients with more severe CF mutations remain significantly more affected. Many factors affect the growth hormone-IGF-1 axis and the growth plate of the long bones, including the chronic inflammatory state associated with CF. There are also increasing data on the direct effects of CFTR on bone and implications for CFTR modulators in attaining optimal growth. Treatments aimed at improving growth in CF are also reviewed here.
Keywords: Cystic fibrosis; Growth; Growth delay; Short stature.
Copyright © 2019 European Cystic Fibrosis Society. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Beker LT, Russek-Cohen E, Fink RJ. Stature as a prognostic factor in cystic fibrosis survival. J Am Diet Assoc. 2001;101(4):438–42. - PubMed
-
- Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, et al. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr. 2003;142(6):624–30. - PubMed
-
- McColley SA, Schechter MS, Morgan WJ, Pasta DJ, Craib ML, Konstan MW. Risk factors for mortality before age 18 years in cystic fibrosis. Pediatr Pulmonol. 2017;52(7):909–15. - PubMed
-
- Yen EH, Quinton H, Borowitz D. Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. J Pediatr. 2013;162(3):530–5 e1. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
