

The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism
- PMID: 31686003
- PMCID: PMC7314312
- DOI: 10.1038/s41568-019-0216-7
The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism
Abstract
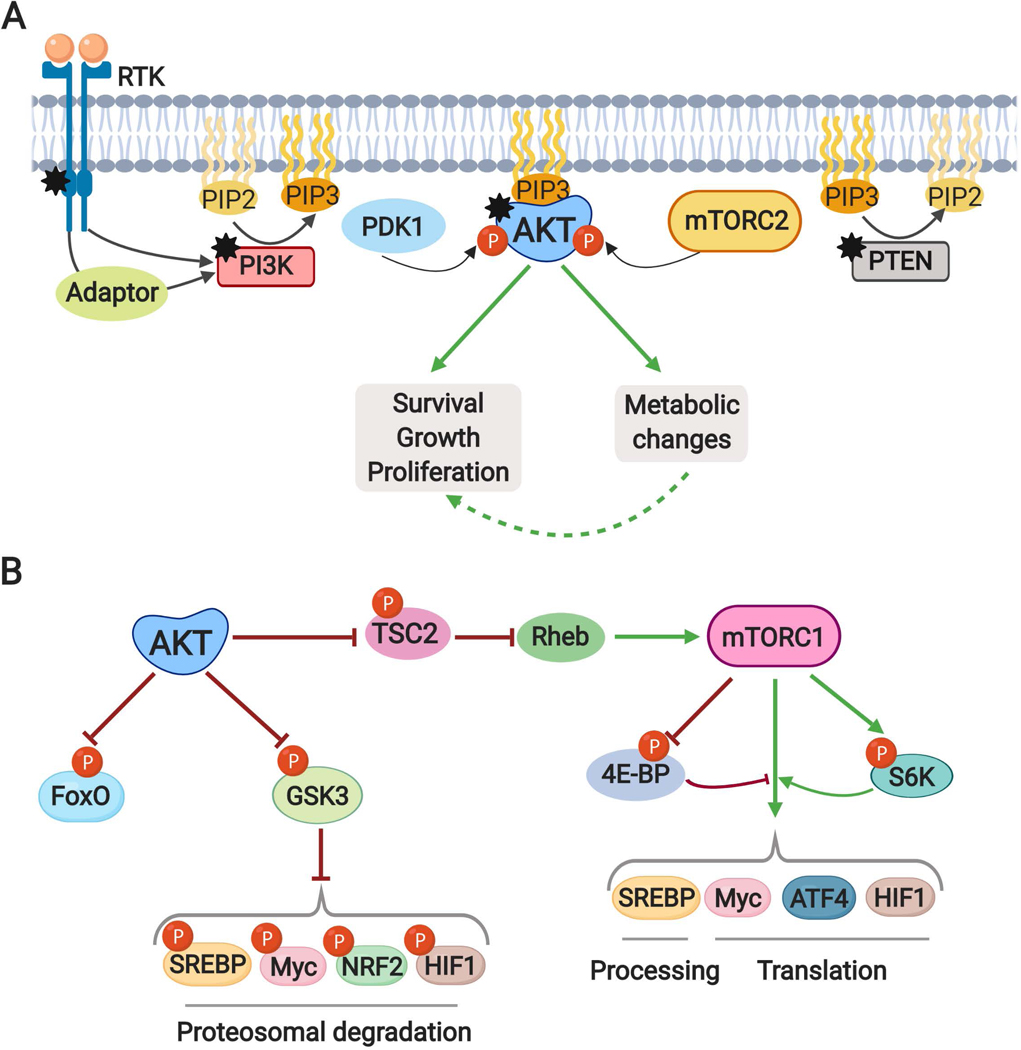
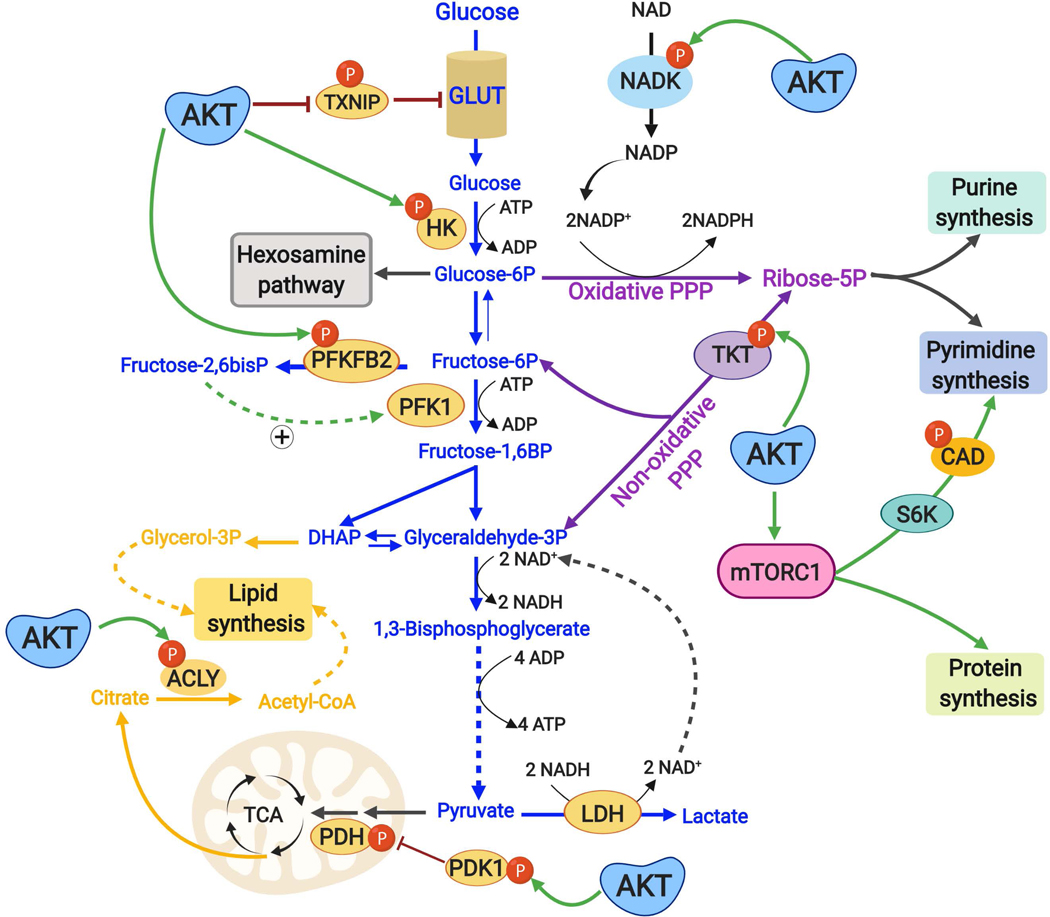
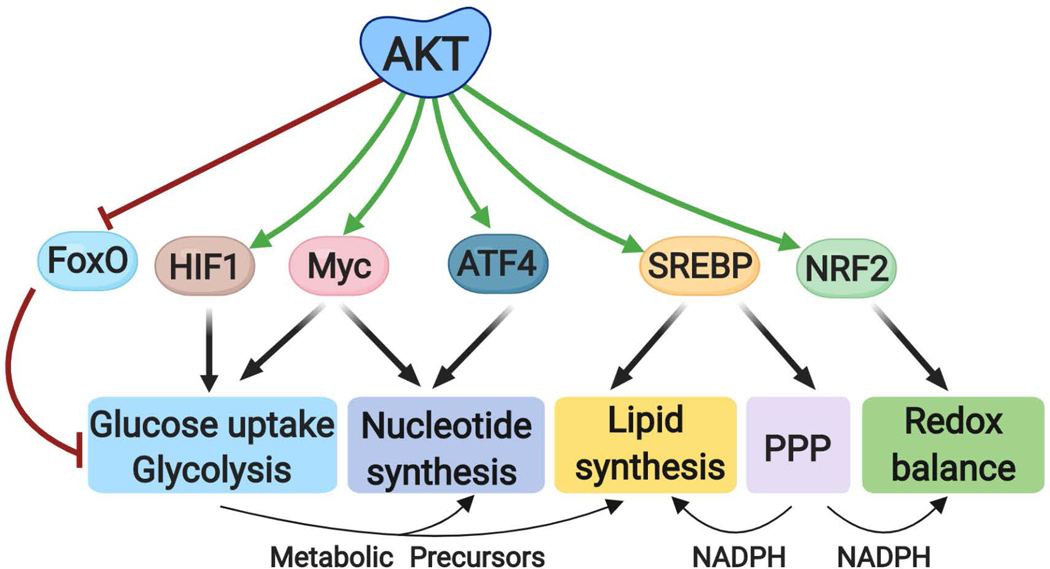
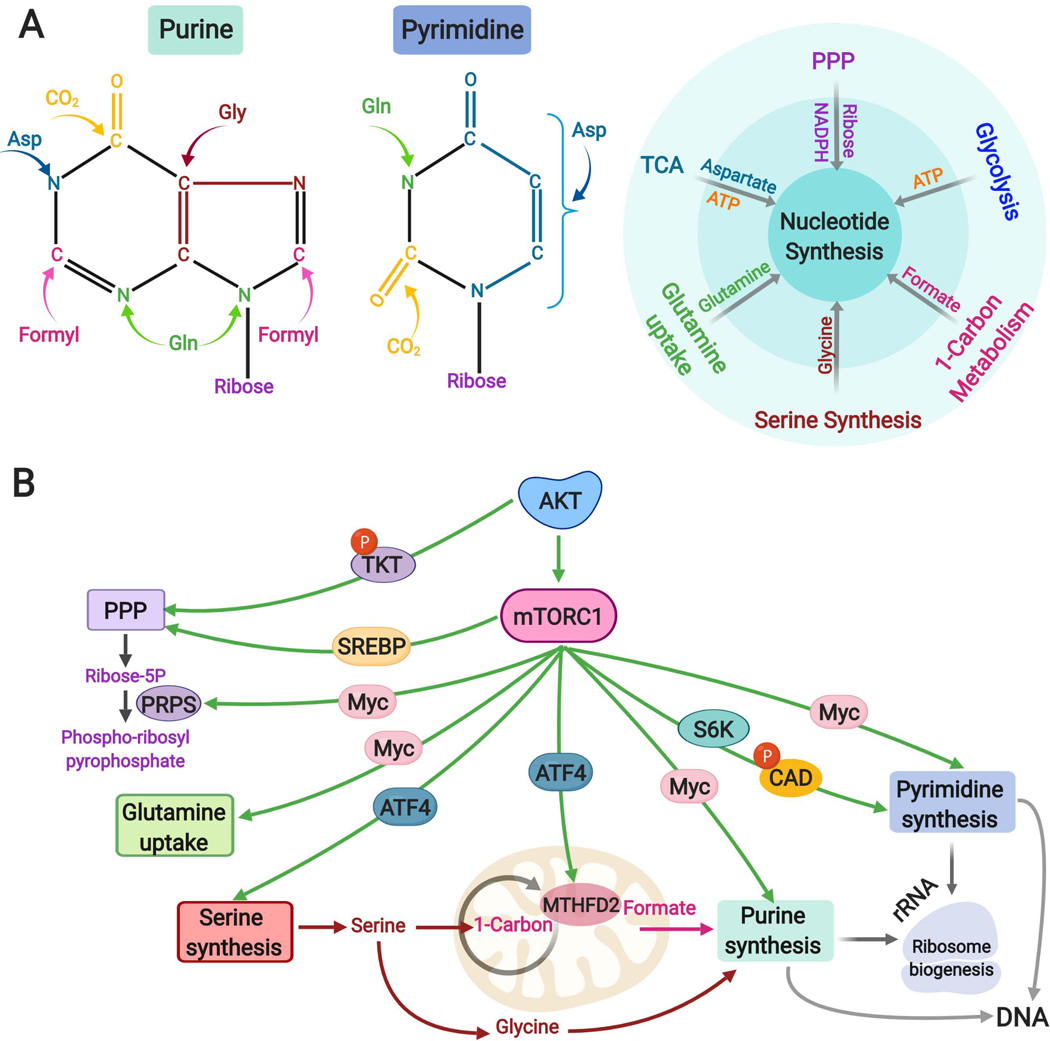
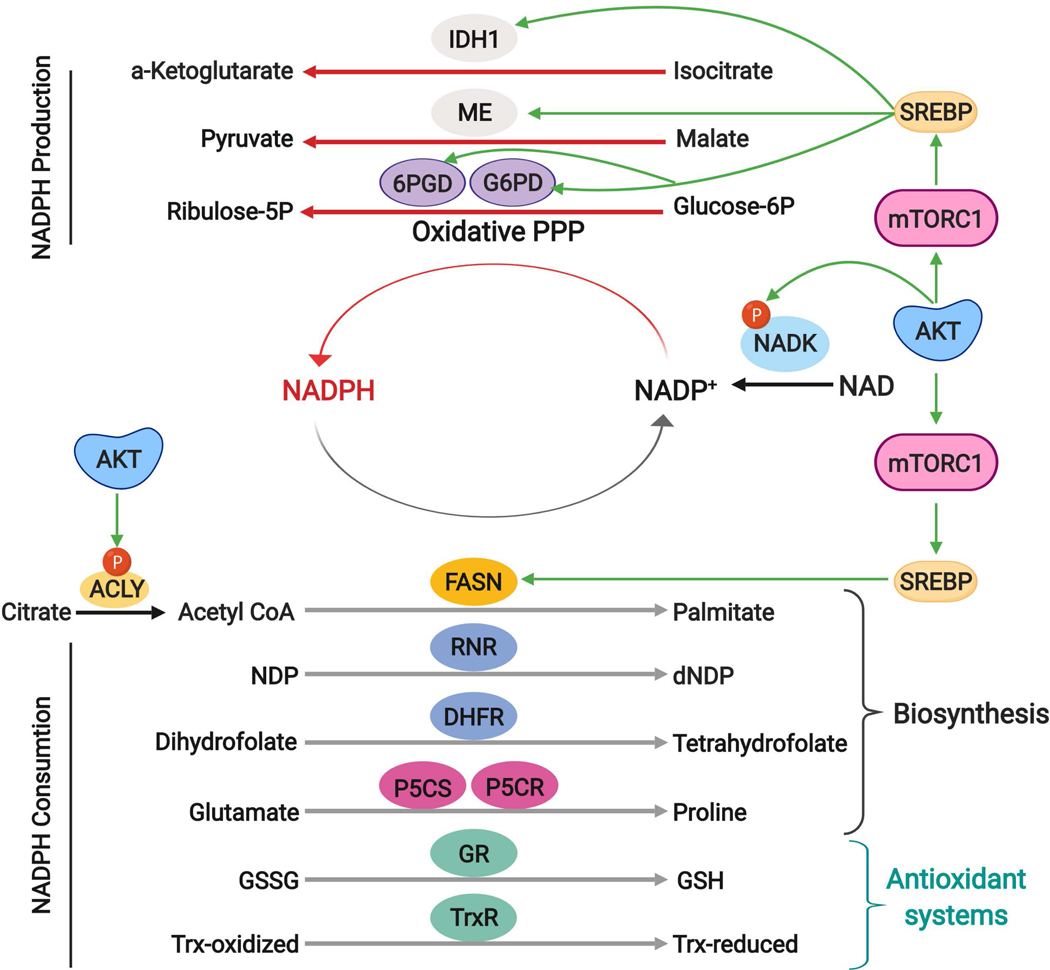
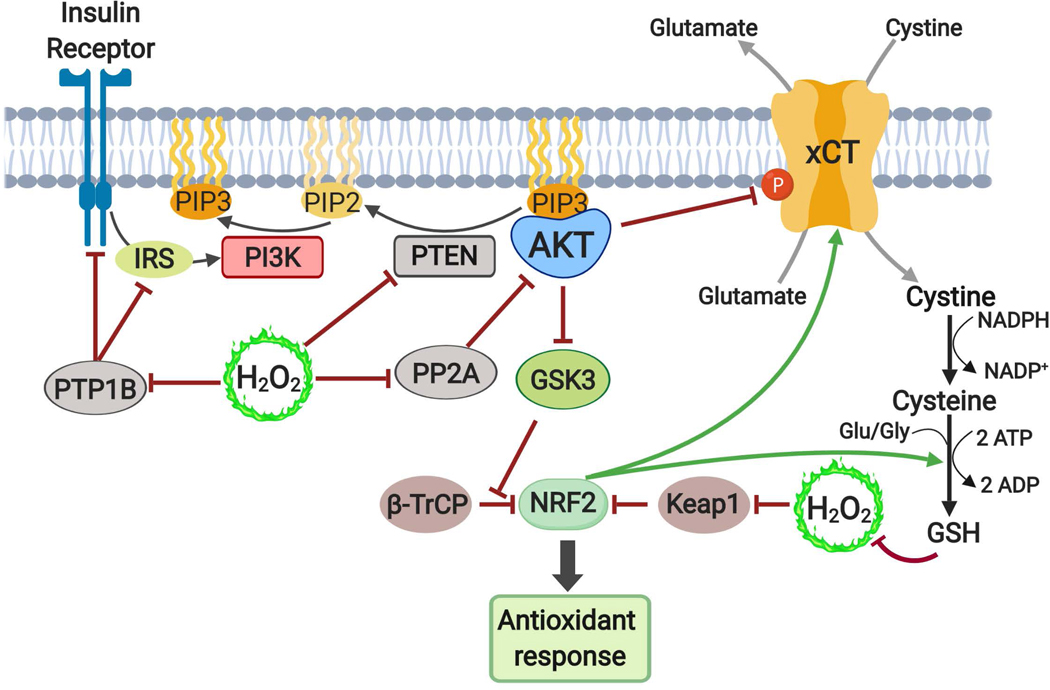
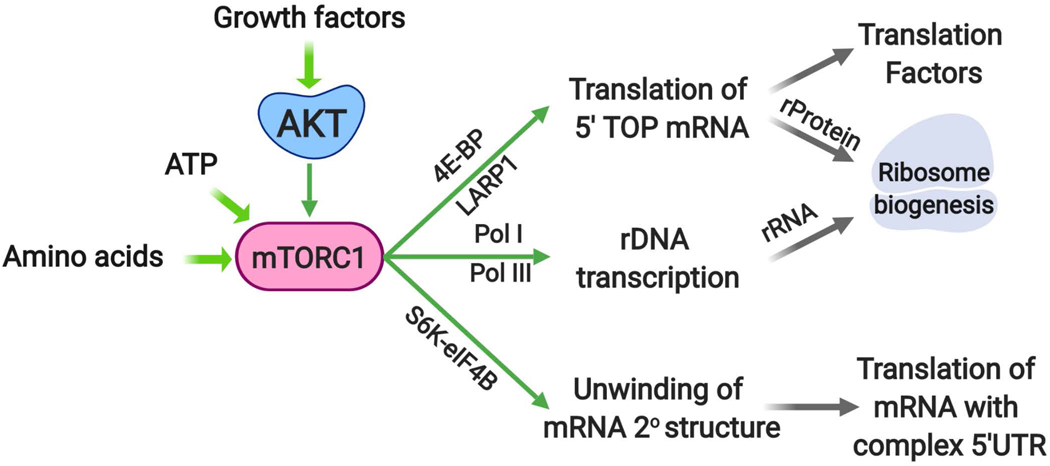
The altered metabolic programme of cancer cells facilitates their cell-autonomous proliferation and survival. In normal cells, signal transduction pathways control core cellular functions, including metabolism, to couple the signals from exogenous growth factors, cytokines or hormones to adaptive changes in cell physiology. The ubiquitous, growth factor-regulated phosphoinositide 3-kinase (PI3K)-AKT signalling network has diverse downstream effects on cellular metabolism, through either direct regulation of nutrient transporters and metabolic enzymes or the control of transcription factors that regulate the expression of key components of metabolic pathways. Aberrant activation of this signalling network is one of the most frequent events in human cancer and serves to disconnect the control of cell growth, survival and metabolism from exogenous growth stimuli. Here we discuss our current understanding of the molecular events controlling cellular metabolism downstream of PI3K and AKT and of how these events couple two major hallmarks of cancer: growth factor independence through oncogenic signalling and metabolic reprogramming to support cell survival and proliferation.
Figures
References
-
- Alessi DR et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7, 261–269 (1997). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
