

Hybrid Electron Microscopy Normal Mode Analysis with Scipion
- PMID: 31693263
- PMCID: PMC6933837
- DOI: 10.1002/pro.3772
Hybrid Electron Microscopy Normal Mode Analysis with Scipion
Abstract
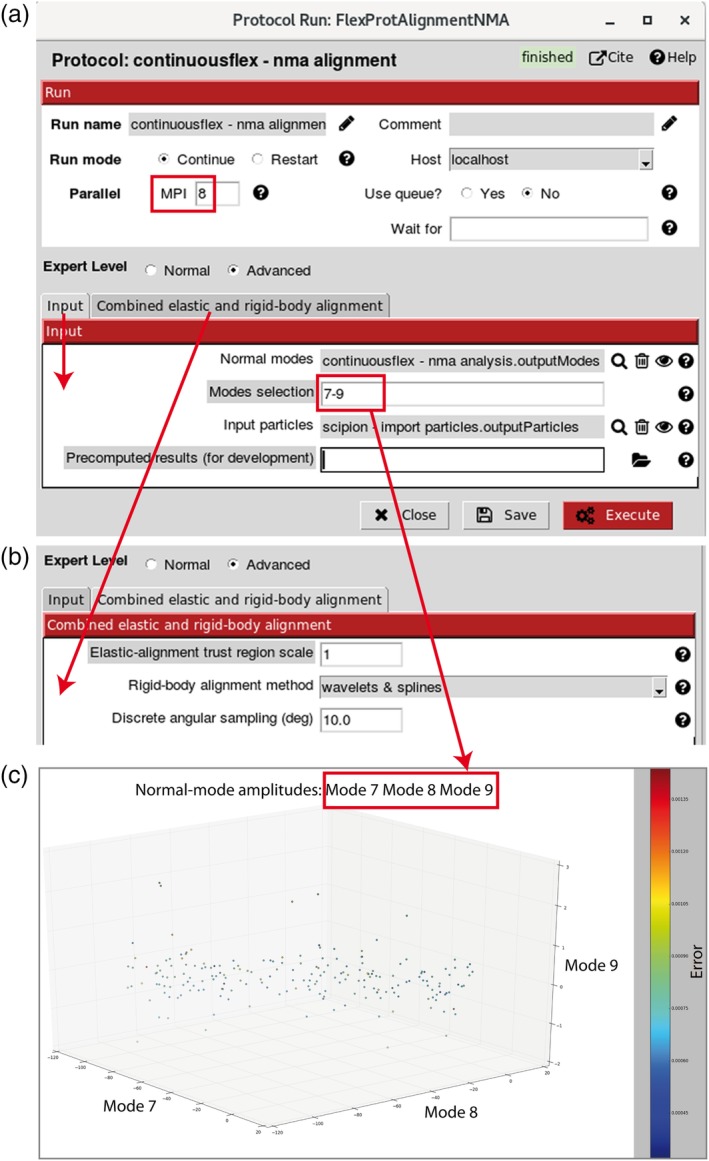
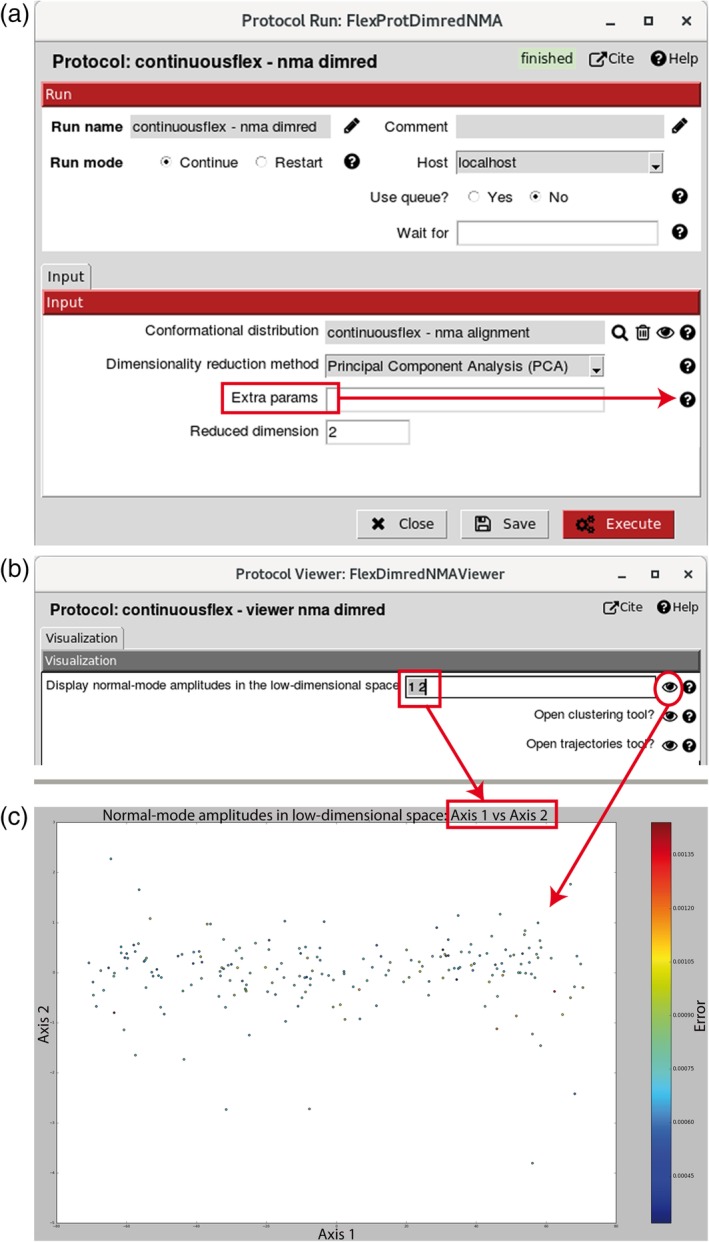
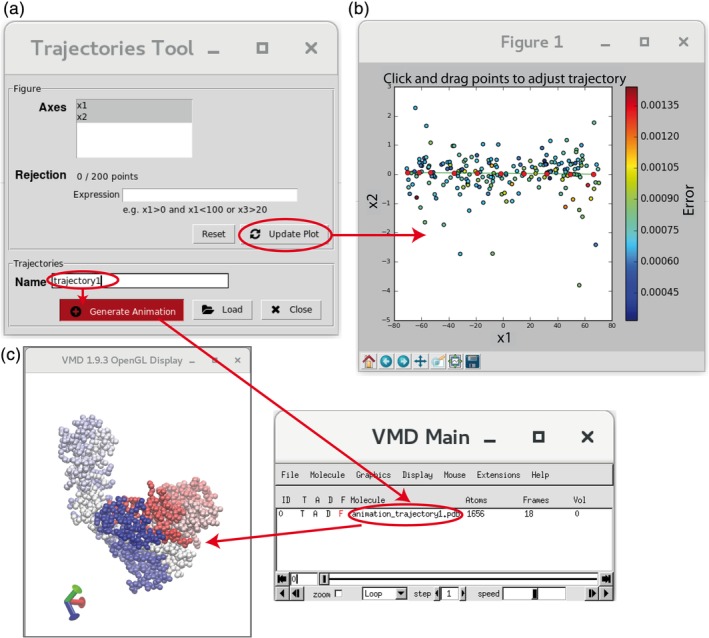
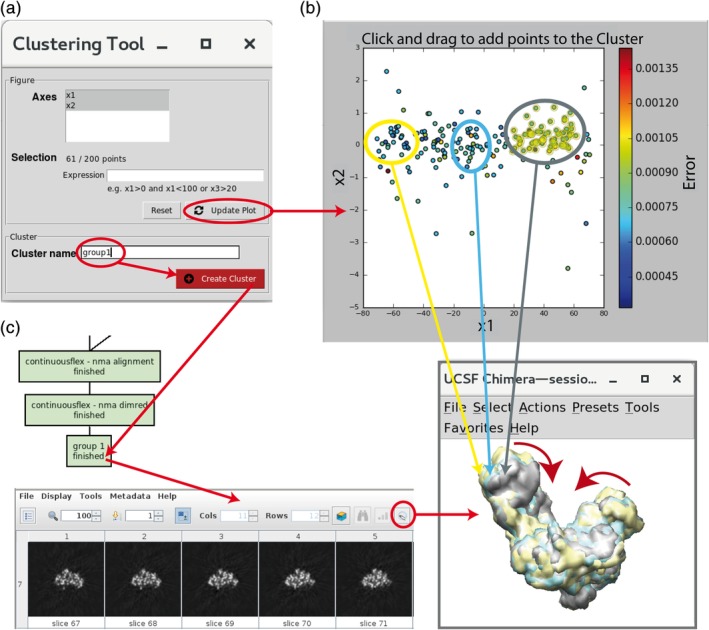
Hybrid Electron Microscopy Normal Mode Analysis (HEMNMA) method was introduced in 2014. HEMNMA computes normal modes of a reference model (an atomic structure or an electron microscopy map) of a molecular complex and uses this model and its normal modes to analyze single-particle images of the complex to obtain information on its continuous conformational changes, by determining the full distribution of conformational variability from the images. An advantage of HEMNMA is a simultaneous determination of all parameters of each image (particle conformation, orientation, and shift) through their iterative optimization, which allows applications of HEMNMA even when the effects of conformational changes dominate those of orientational changes. HEMNMA was first implemented in Xmipp and was using MATLAB for statistical analysis of obtained conformational distributions and for fitting of underlying trajectories of conformational changes. A HEMNMA implementation independent of MATLAB is now available as part of a plugin of Scipion V2.0 (http://scipion.i2pc.es). This plugin, named ContinuousFlex, can be installed by following the instructions at https://pypi.org/project/scipion-em-continuousflex. In this article, we present this new HEMNMA software, which is user-friendly, totally free, and open-source. STATEMENT FOR A BROADER AUDIENCE: This article presents Hybrid Electron Microscopy Normal Mode Analysis (HEMNMA) software that allows analyzing single-particle images of a complex to obtain information on continuous conformational changes of the complex, by determining the full distribution of conformational variability from the images. The HEMNMA software is user-friendly, totally free, open-source, and available as part of ContinuousFlex plugin (https://pypi.org/project/scipion-em-continuousflex) of Scipion V2.0 (http://scipion.i2pc.es).
Keywords: continuous conformational changes; cryo-electron microscopy; dynamics; normal mode analysis; single-particle analysis; software; structure.
© 2019 The Protein Society.
Figures
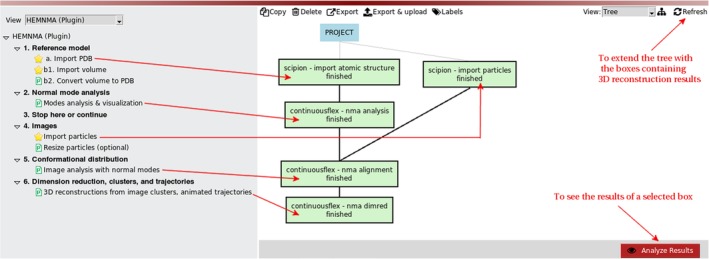
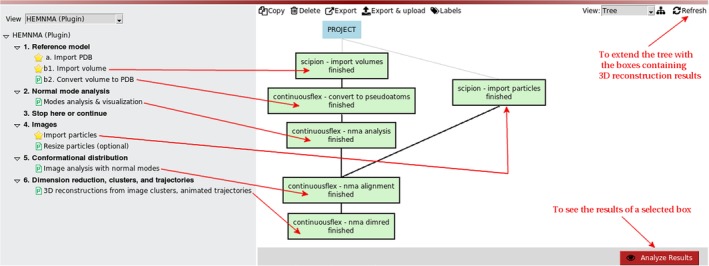
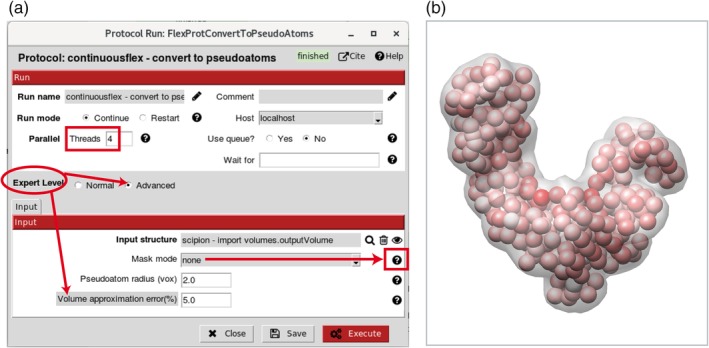
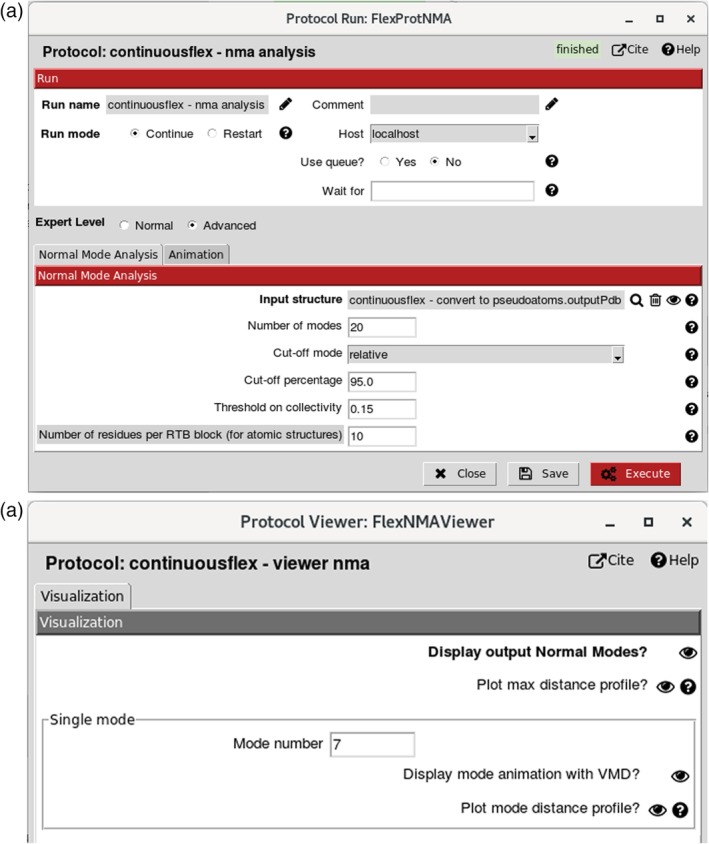
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
