

Mitochondria: at the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease
- PMID: 31693390
- PMCID: PMC6985875
- DOI: 10.1152/ajplung.00329.2019
Mitochondria: at the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease
Abstract
Disturbances in mitochondrial structure and function in lung epithelial cells have been implicated in the pathogenesis of various lung diseases, including chronic obstructive pulmonary disease (COPD). Such disturbances affect not only cellular energy metabolism but also alter a range of indispensable cellular homeostatic functions in which mitochondria are known to be involved. These range from cellular differentiation, cell death pathways, and cellular remodeling to physical barrier function and innate immunity, all of which are known to be impacted by exposure to cigarette smoke and have been linked to COPD pathogenesis. Next to their well-established role as the first physical frontline against external insults, lung epithelial cells are immunologically active. Malfunctioning epithelial cells with defective mitochondria are unable to maintain homeostasis and respond adequately to further stress or injury, which may ultimately shape the phenotype of lung diseases. In this review, we provide a comprehensive overview of the impact of cigarette smoke on the development of mitochondrial dysfunction in the lung epithelium and highlight the consequences for cell function, innate immune responses, epithelial remodeling, and epithelial barrier function in COPD. We also discuss the applicability and potential therapeutic value of recently proposed strategies for the restoration of mitochondrial function in the treatment of COPD.
Keywords: COPD; cigarette smoke; lung epithelial cells; mitochondrial dysfunction.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
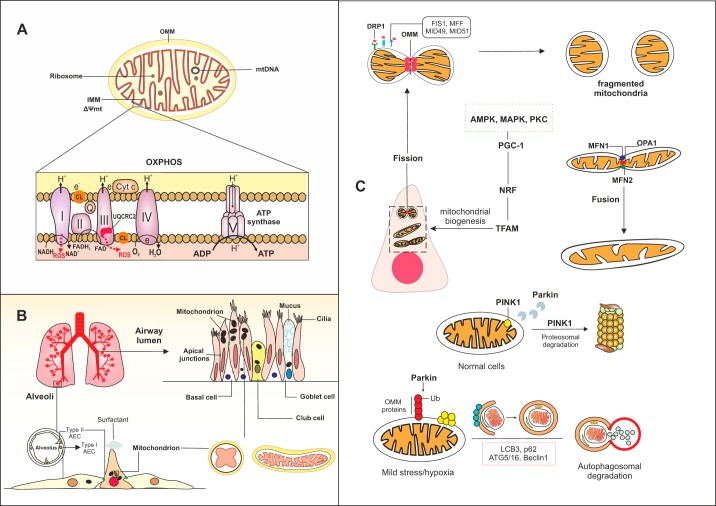
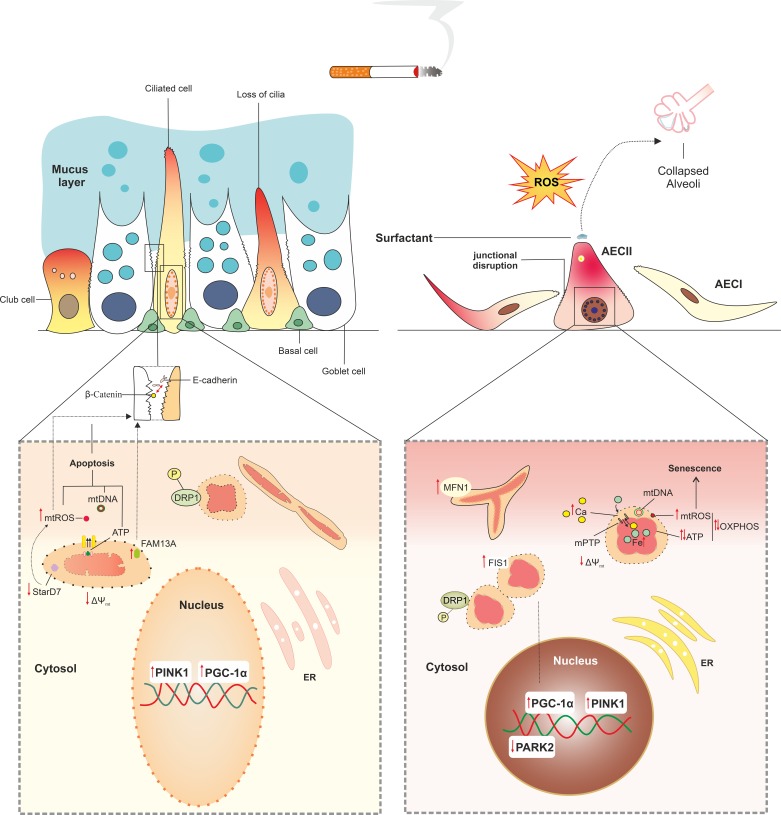
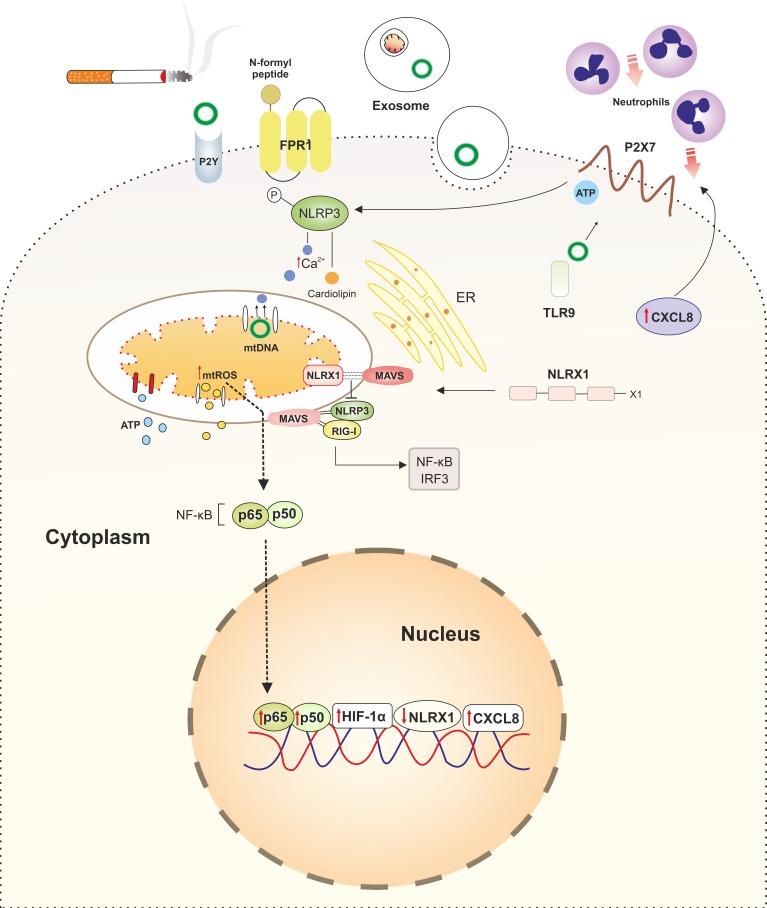
References
-
- Agarwal AR, Zhao L, Sancheti H, Sundar IK, Rahman I, Cadenas E. Short-term cigarette smoke exposure induces reversible changes in energy metabolism and cellular redox status independent of inflammatory responses in mouse lungs. Am J Physiol Lung Cell Mol Physiol 303: L889–L898, 2012. doi: 10.1152/ajplung.00219.2012. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical