

Autoimmunity as a continuum in primary immunodeficiency
- PMID: 31693597
- PMCID: PMC6919226
- DOI: 10.1097/MOP.0000000000000833
Autoimmunity as a continuum in primary immunodeficiency
Abstract
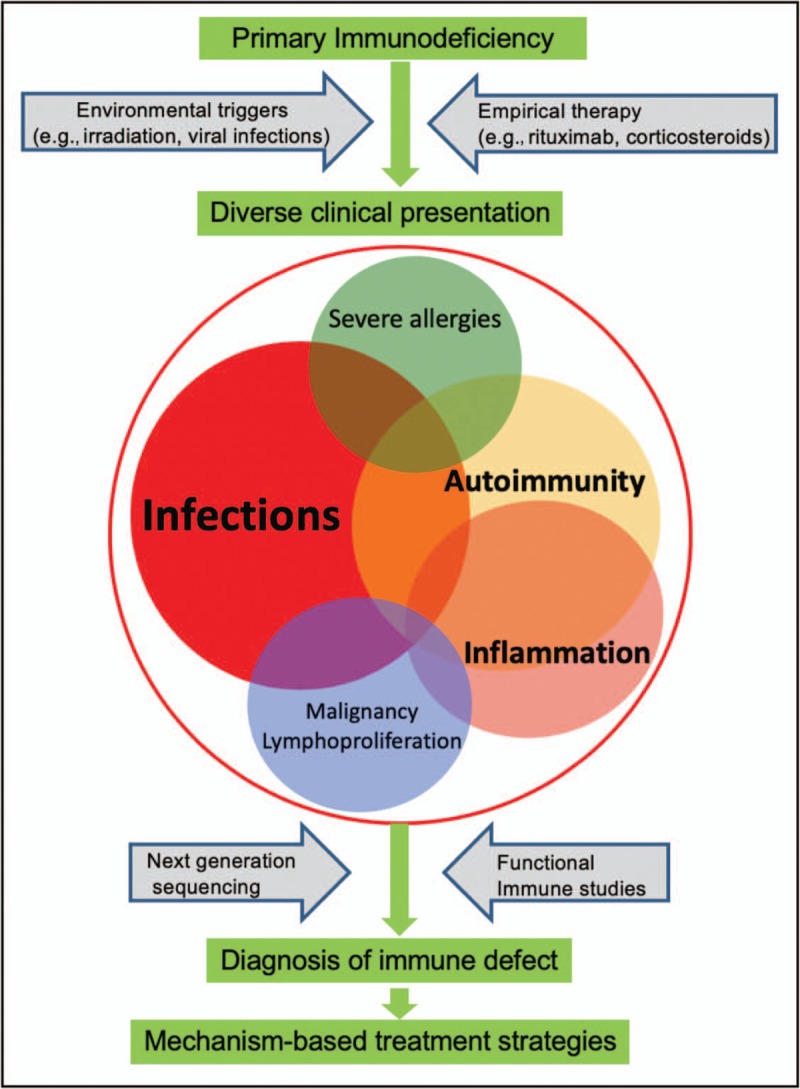
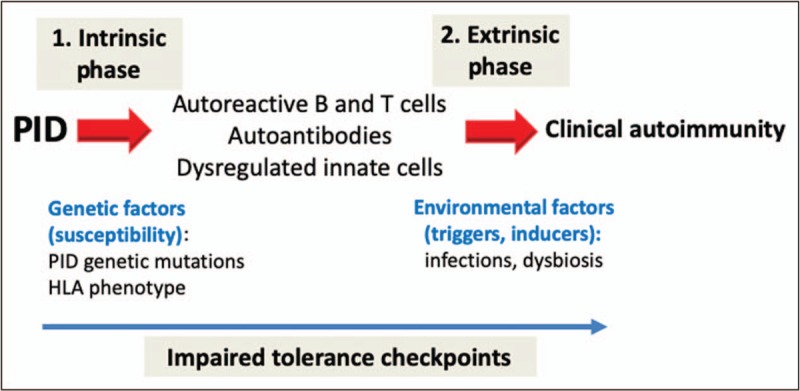
Purpose of review: Primary immunodeficiency disorders (PIDs) are no longer defined by infections alone. First clinical sign or sequelae of PID may include autoimmunity, such as cytopenias, arthritis or enteropathy. This review addresses the latest in multidisciplinary approaches for expanding clinical phenotypes of PIDs with autoimmunity, including new presentations of known entities and novel gene defects. We also discuss diagnostic tools for identifying the distinct changes in immune cells subsets and autoantibodies, mechanistic understanding of the process, and targeted treatment and indications for hematopoietic stem-cell transplantation (HSCT).
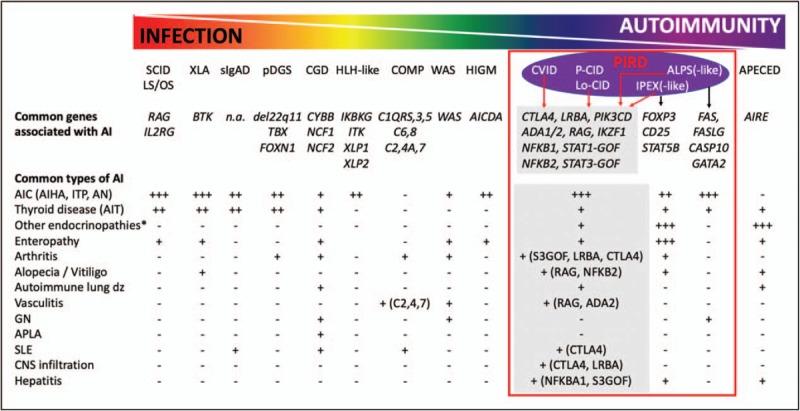
Recent findings: In the past years, increased awareness and use of genetic screening, confirmatory functional studies and immunological biomarkers opened the door for early recognition of PIDs among patients with autoimmunity. Large cohort studies detail the clinical spectrum and treatment outcome of PIDs with autoimmunity with specific immune genes (e.g., CTLA4, LRBA, PI3Kδ, NFKB1, RAG). The benefit of early recognition is initiation of targeted therapies with precise re-balancing of the dysregulated immune pathways (e.g., biologicals) or definitive therapy (e.g., HSCT).
Summary: Clinical presentation of patients with PID and autoimmunity is highly variable and requires in-depth diagnostics and precision medicine approaches.
Figures

References
-
- Nagamine K, Peterson P, Scott HS, et al. Positional cloning of the APECED gene. Nat Genet 1997; 17:393–398. - PubMed
-
- Finnish-German AC. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 1997; 17:399–403. - PubMed
-
- Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27:20–21. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
