

Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design
- PMID: 31693904
- PMCID: PMC6880934
- DOI: 10.1016/j.celrep.2019.09.071
Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design
Abstract
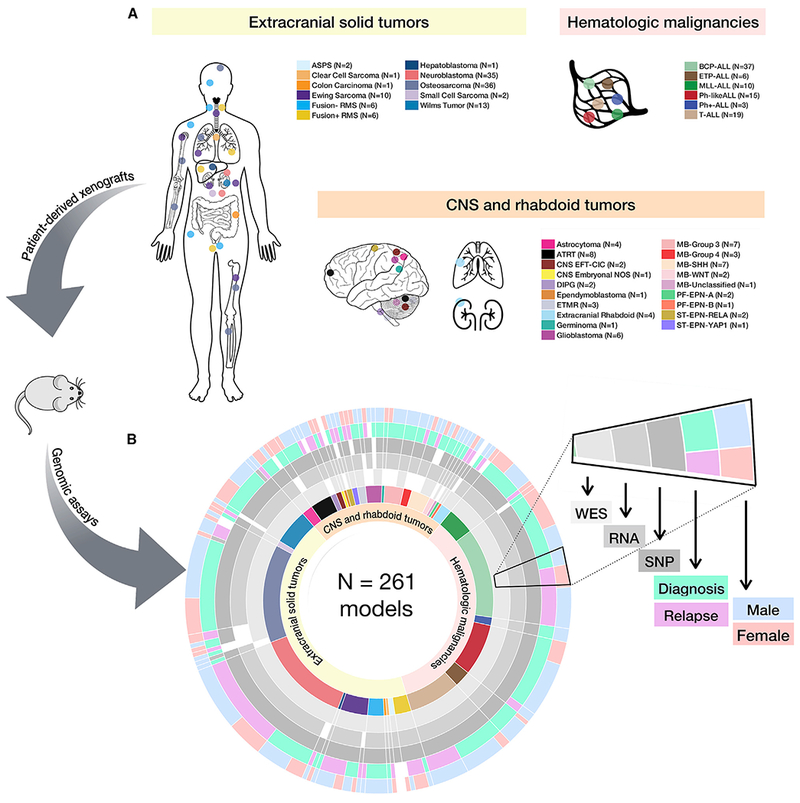
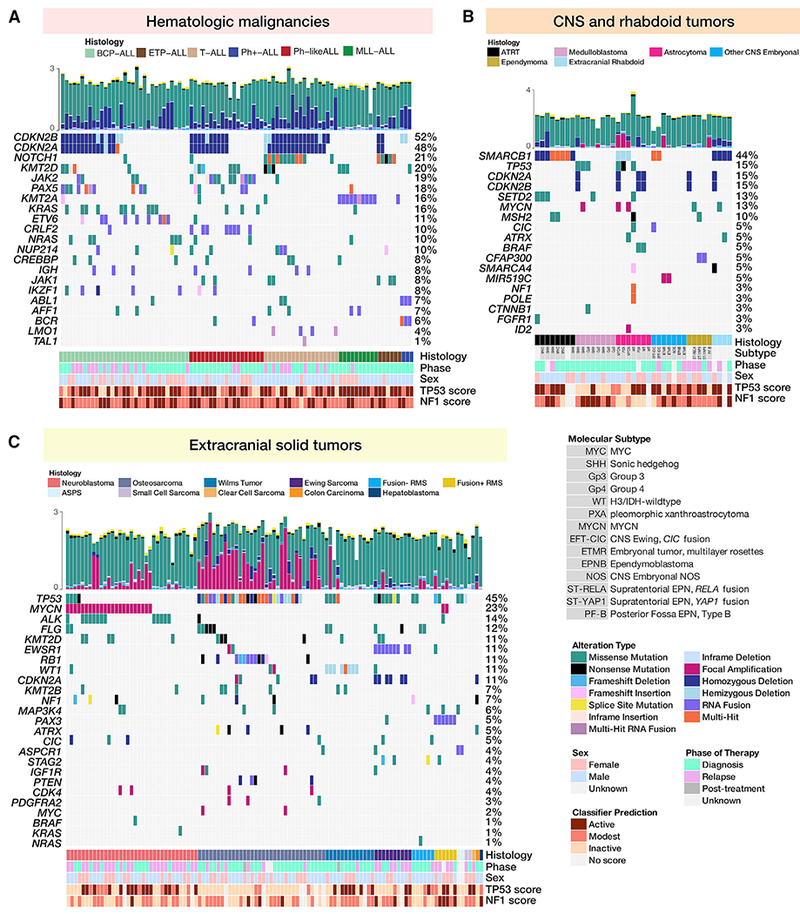
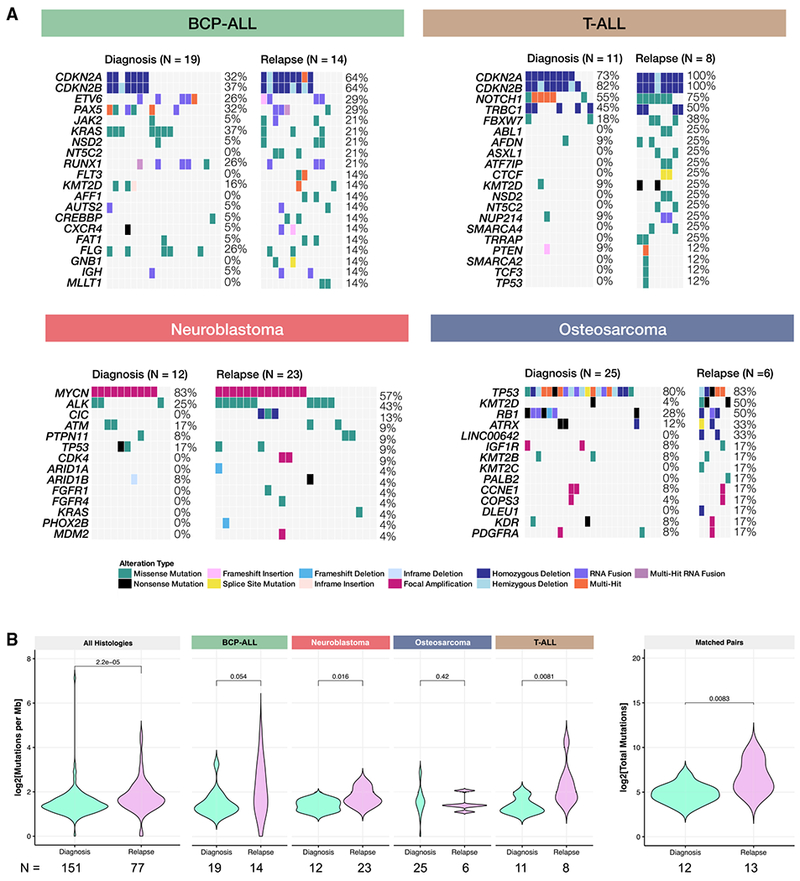
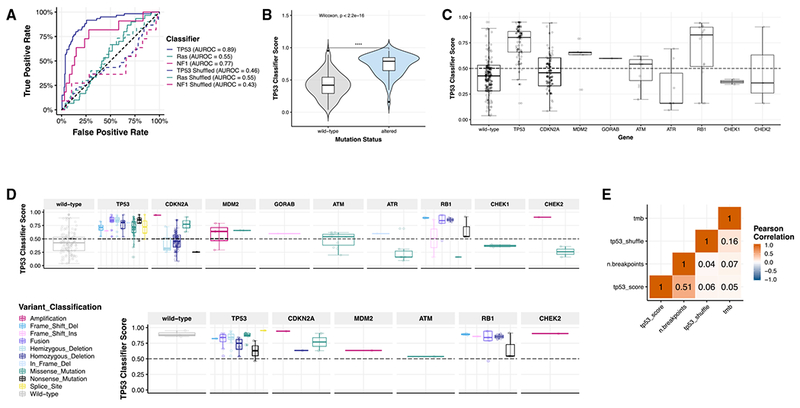
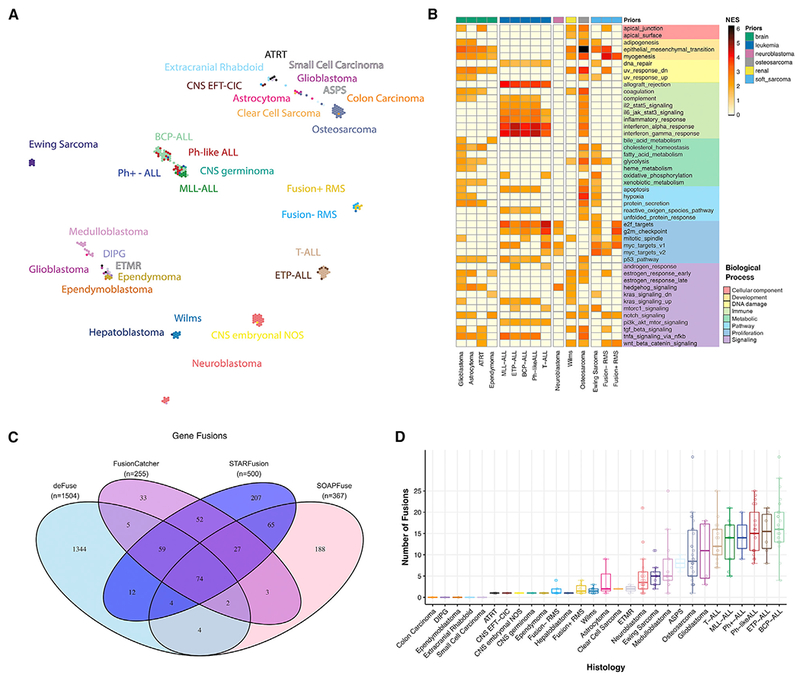
Accelerating cures for children with cancer remains an immediate challenge as a result of extensive oncogenic heterogeneity between and within histologies, distinct molecular mechanisms evolving between diagnosis and relapsed disease, and limited therapeutic options. To systematically prioritize and rationally test novel agents in preclinical murine models, researchers within the Pediatric Preclinical Testing Consortium are continuously developing patient-derived xenografts (PDXs)-many of which are refractory to current standard-of-care treatments-from high-risk childhood cancers. Here, we genomically characterize 261 PDX models from 37 unique pediatric cancers; demonstrate faithful recapitulation of histologies and subtypes; and refine our understanding of relapsed disease. In addition, we use expression signatures to classify tumors for TP53 and NF1 pathway inactivation. We anticipate that these data will serve as a resource for pediatric oncology drug development and will guide rational clinical trial design for children with cancer.
Keywords: classifier; copy number profiling; patient-derived xenograft; pediatric cancer; preclinical testing; relapse; transcriptome sequencing; whole-exome sequencing.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
References
-
- American Childhood Cancer Organization (2014). Special Section: Cancer in Children & Adolescents In Cancer Facts and Figures 2014 (The American Cancer Society), pp. 25–42.
-
- Baker SC, Bauer SR, Beyer RP, Brenton JD, Bromley B, Burrill J, Causton H, Conley MP, Elespuru R, Fero M, et al. ; External RNA Controls Consortium (2005). The External RNA Controls Consortium: a progress report. Nat. Methods 2, 731–734. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 CA199288/CA/NCI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- U01 CA199000/CA/NCI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- U01 CA199287/CA/NCI NIH HHS/United States
- R35 CA220500/CA/NCI NIH HHS/United States
- U01 CA199222/CA/NCI NIH HHS/United States
- U01 CA199221/CA/NCI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- R01 CA221957/CA/NCI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- L40 CA220942/CA/NCI NIH HHS/United States
- U01 CA263981/CA/NCI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- T32 CA009615/CA/NCI NIH HHS/United States
- U01 CA199297/CA/NCI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- R01 NS095411/NS/NINDS NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
