

From cytogenetics to cytogenomics: whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability
- PMID: 31694722
- PMCID: PMC6836550
- DOI: 10.1186/s13073-019-0675-1
From cytogenetics to cytogenomics: whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability
Abstract
Background: Since different types of genetic variants, from single nucleotide variants (SNVs) to large chromosomal rearrangements, underlie intellectual disability, we evaluated the use of whole-genome sequencing (WGS) rather than chromosomal microarray analysis (CMA) as a first-line genetic diagnostic test.
Methods: We analyzed three cohorts with short-read WGS: (i) a retrospective cohort with validated copy number variants (CNVs) (cohort 1, n = 68), (ii) individuals referred for monogenic multi-gene panels (cohort 2, n = 156), and (iii) 100 prospective, consecutive cases referred to our center for CMA (cohort 3). Bioinformatic tools developed include FindSV, SVDB, Rhocall, Rhoviz, and vcf2cytosure.
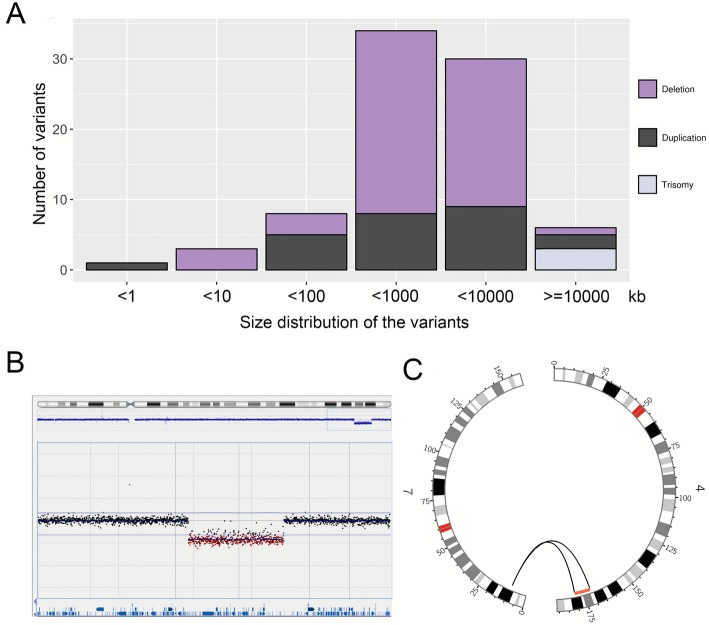
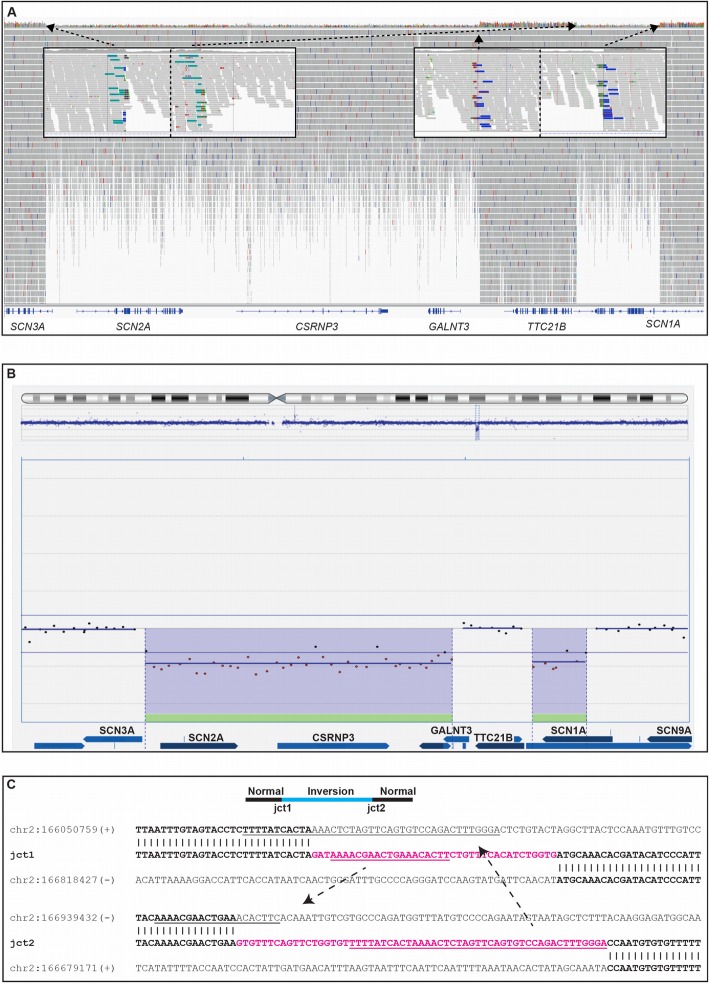
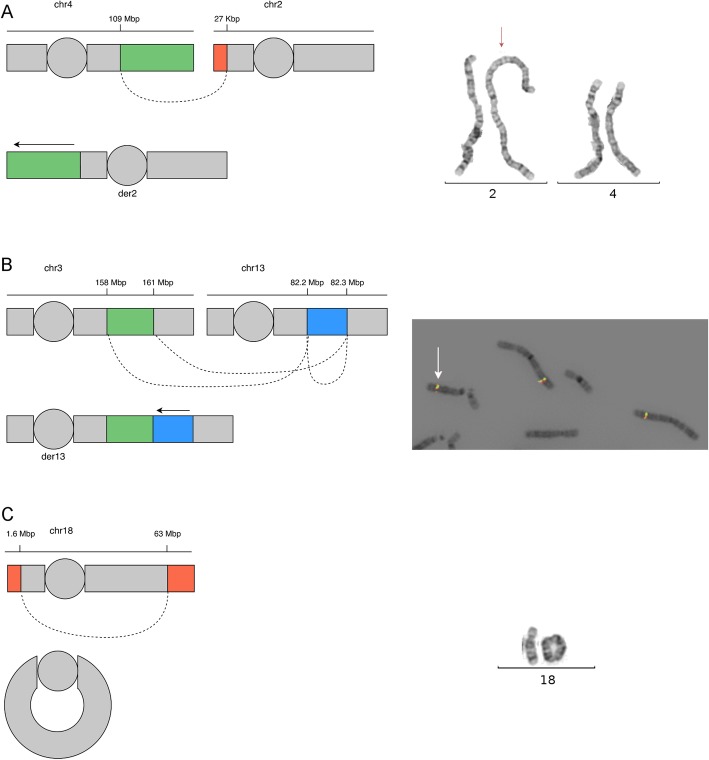
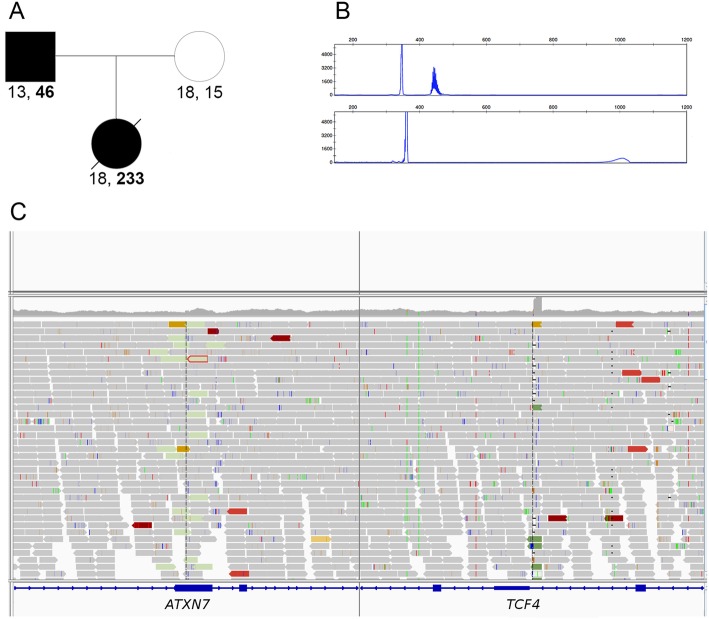
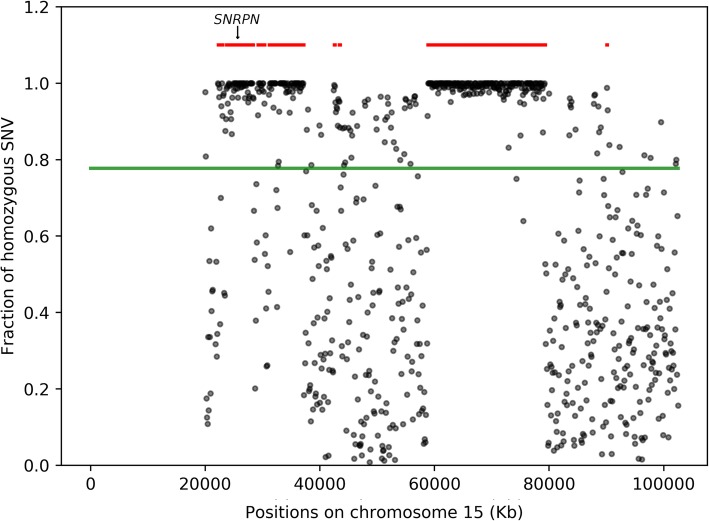
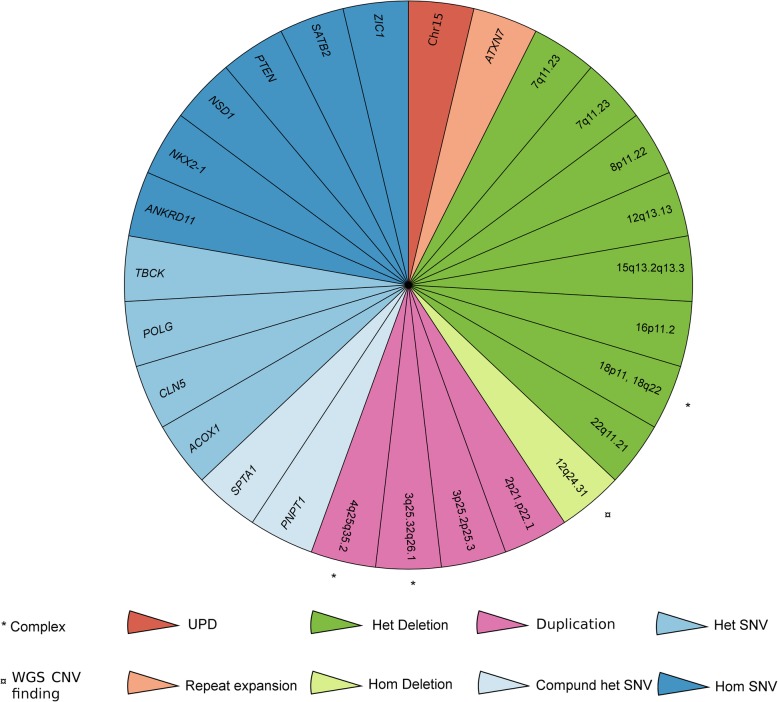
Results: First, we validated our structural variant (SV)-calling pipeline on cohort 1, consisting of three trisomies and 79 deletions and duplications with a median size of 850 kb (min 500 bp, max 155 Mb). All variants were detected. Second, we utilized the same pipeline in cohort 2 and analyzed with monogenic WGS panels, increasing the diagnostic yield to 8%. Next, cohort 3 was analyzed by both CMA and WGS. The WGS data was processed for large (> 10 kb) SVs genome-wide and for exonic SVs and SNVs in a panel of 887 genes linked to intellectual disability as well as genes matched to patient-specific Human Phenotype Ontology (HPO) phenotypes. This yielded a total of 25 pathogenic variants (SNVs or SVs), of which 12 were detected by CMA as well. We also applied short tandem repeat (STR) expansion detection and discovered one pathologic expansion in ATXN7. Finally, a case of Prader-Willi syndrome with uniparental disomy (UPD) was validated in the WGS data. Important positional information was obtained in all cohorts. Remarkably, 7% of the analyzed cases harbored complex structural variants, as exemplified by a ring chromosome and two duplications found to be an insertional translocation and part of a cryptic unbalanced translocation, respectively.
Conclusion: The overall diagnostic rate of 27% was more than doubled compared to clinical microarray (12%). Using WGS, we detected a wide range of SVs with high accuracy. Since the WGS data also allowed for analysis of SNVs, UPD, and STRs, it represents a powerful comprehensive genetic test in a clinical diagnostic laboratory setting.
Keywords: Copy number variation; Intellectual disability; Monogenic disease; Repeat expansion; Single nucleotide variant; Structural variation; Uniparental disomy; Whole-genome sequencing.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
