

The Positive Allosteric Modulator of α 2/3-Containing GABAA Receptors, KRM-II-81, Is Active in Pharmaco-Resistant Models of Epilepsy and Reduces Hyperexcitability after Traumatic Brain Injury
- PMID: 31694876
- PMCID: PMC6927408
- DOI: 10.1124/jpet.119.260968
The Positive Allosteric Modulator of α 2/3-Containing GABAA Receptors, KRM-II-81, Is Active in Pharmaco-Resistant Models of Epilepsy and Reduces Hyperexcitability after Traumatic Brain Injury
Abstract
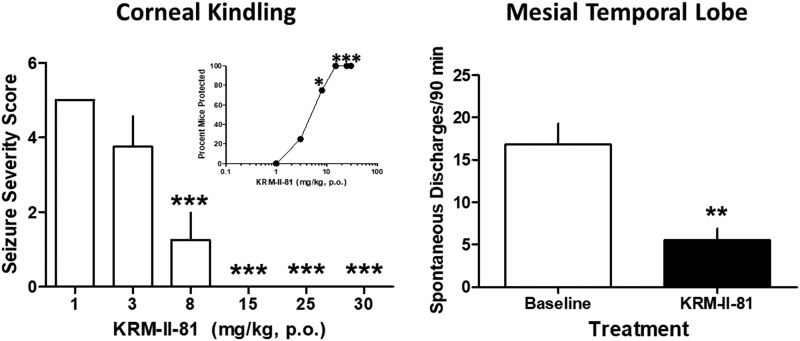
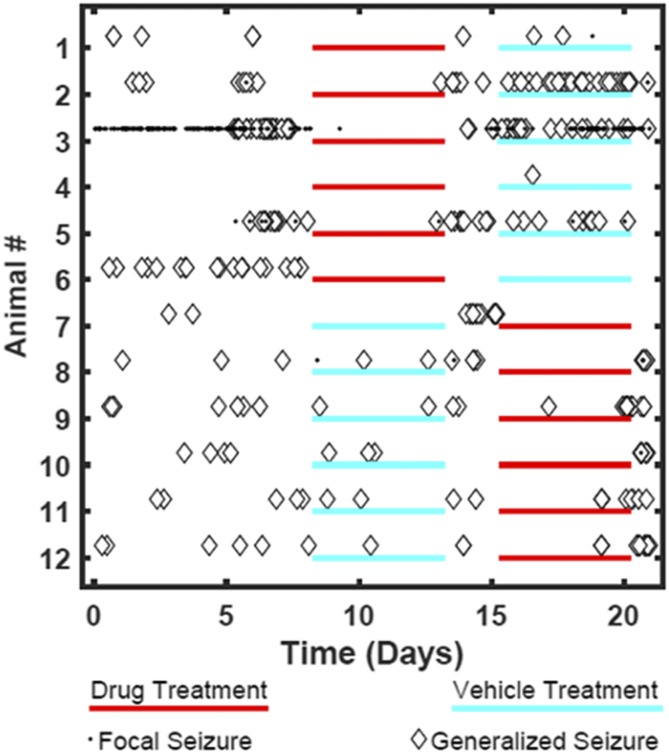
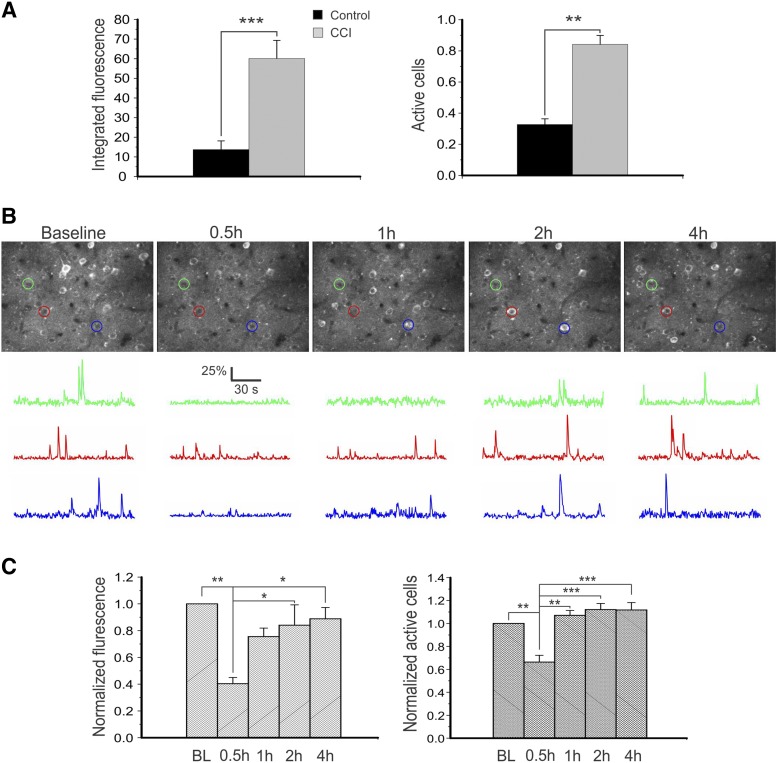
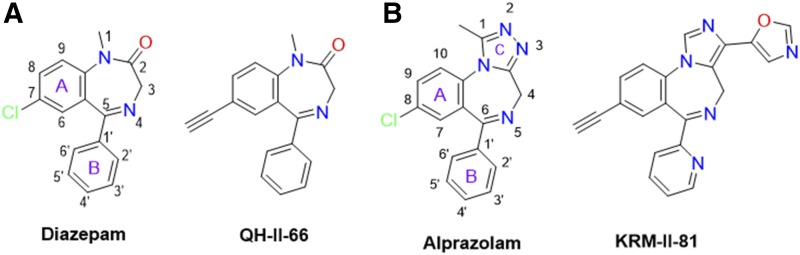
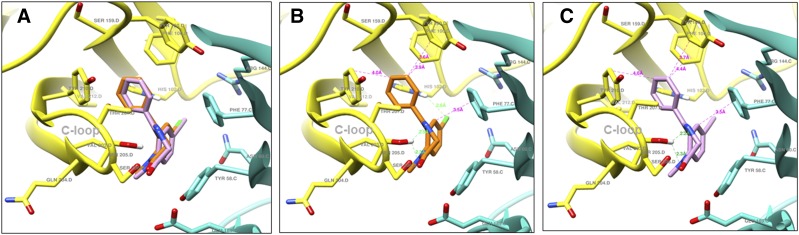
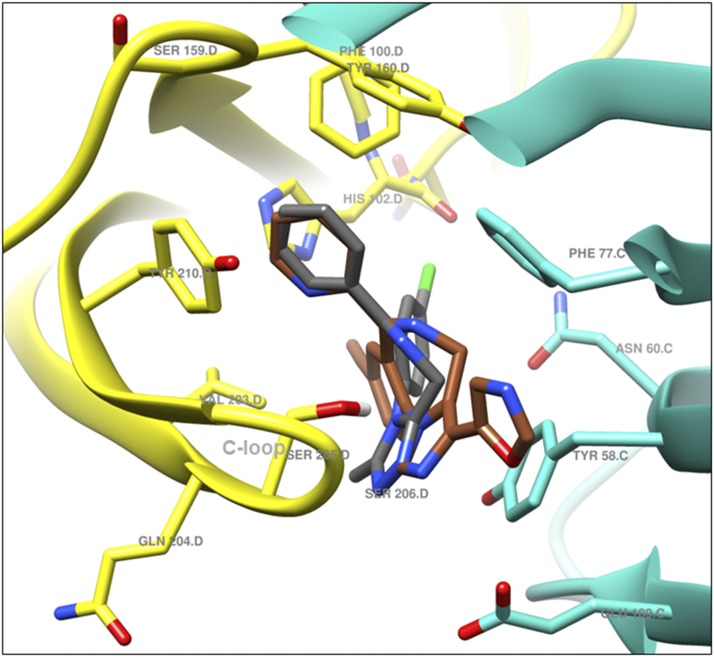
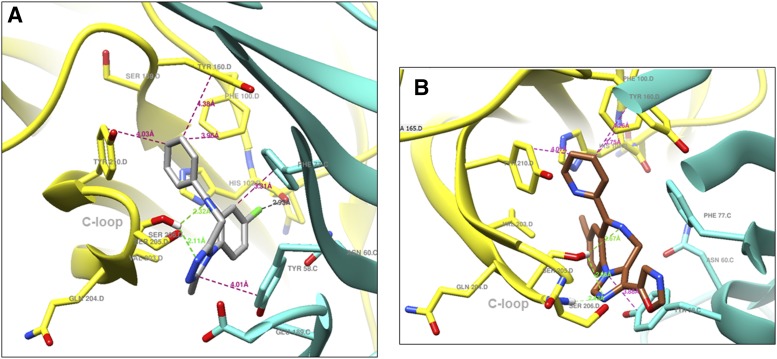
The imidizodiazepine, 5-(8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)oxazole (KRM-II-81), is selective for α2/3-containing GABAA receptors. KRM-II-81 dampens seizure activity in rodent models with enhanced efficacy and reduced motor-impairment compared with diazepam. In the present study, KRM-II-81 was studied in assays designed to detect antiepileptics with improved chances of impacting pharmaco-resistant epilepsies. The potential for reducing neural hyperactivity weeks after traumatic brain injury was also studied. KRM-II-81 suppressed convulsions in corneal-kindled mice. Mice with kainate-induced mesial temporal lobe seizures exhibited spontaneous recurrent hippocampal paroxysmal discharges that were significantly reduced by KRM-II-81 (15 mg/kg, orally). KRM-II-81 also decreased convulsions in rats undergoing amygdala kindling in the presence of lamotrigine (lamotrigine-insensitive model) (ED50 = 19 mg/kg, i.p.). KRM-II-81 reduced focal and generalized seizures in a kainate-induced chronic epilepsy model in rats (20 mg/kg, i.p., three times per day). In mice with damage to the left cerebral cortex by controlled-cortical impact, enduring neuronal hyperactivity was dampened by KRM-II-81 (10 mg/kg, i.p.) as observed through in vivo two-photon imaging of layer II/III pyramidal neurons in GCaMP6-expressing transgenic mice. No notable side effects emerged up to doses of 300 mg/kg KRM-II-81. Molecular modeling studies were conducted: docking in the binding site of the α1β3γ2L GABAA receptor showed that replacing the C8 chlorine atom of alprazolam with the acetylene of KRM-II-81 led to loss of the key interaction with α1His102, providing a structural rationale for its low affinity for α1-containing GABAA receptors compared with benzodiazepines such as alprazolam. Overall, these findings predict that KRM-II-81 has improved therapeutic potential for epilepsy and post-traumatic epilepsy. SIGNIFICANCE STATEMENT: We describe the effects of a relatively new orally bioavailable small molecule in rodent models of pharmaco-resistant epilepsy and traumatic brain injury. KRM-II-81 is more potent and generally more efficacious than standard-of-care antiepileptics. In silico docking experiments begin to describe the structural basis for the relative lack of motor impairment induced by KRM-II-81. KRM-II-81 has unique structural and anticonvulsant effects, predicting its potential as an improved antiepileptic drug and novel therapy for post-traumatic epilepsy.
Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics.
Conflict of interest statement
Some of the authors are patent holders for KRM-II-81 (G.L., L.K.G., F.R., R.J., and J.M.C.). The University of Wisconsin-Milwaukee is the owner of KRM-II-81. J.M.W. is an adjunct faculty member of the University of Wisconsin-Milwaukee and lead biologist on the antiepileptic drug development effort for KRM-II-81.
Figures
Similar articles
-
Bioisosteres of ethyl 8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo [1,5-a][1,4]diazepine-3-carboxylate (HZ-166) as novel alpha 2,3 selective potentiators of GABAA receptors: Improved bioavailability enhances anticonvulsant efficacy.Neuropharmacology. 2018 Jul 15;137:332-343. doi: 10.1016/j.neuropharm.2018.05.006. Epub 2018 May 3. Neuropharmacology. 2018. PMID: 29778948
-
Structural Analogs of the GABAkine KRM-II-81 Are Orally Bioavailable Anticonvulsants without Sedation.J Pharmacol Exp Ther. 2023 Apr;385(1):50-61. doi: 10.1124/jpet.122.001362. Epub 2023 Feb 6. J Pharmacol Exp Ther. 2023. PMID: 36746611 Free PMC article.
-
The α2,3-selective potentiator of GABAA receptors, KRM-II-81, reduces nociceptive-associated behaviors induced by formalin and spinal nerve ligation in rats.Pharmacol Biochem Behav. 2019 May;180:22-31. doi: 10.1016/j.pbb.2019.02.013. Epub 2019 Feb 27. Pharmacol Biochem Behav. 2019. PMID: 30825491 Free PMC article.
-
The imidazodiazepine, KRM-II-81: An example of a newly emerging generation of GABAkines for neurological and psychiatric disorders.Pharmacol Biochem Behav. 2022 Feb;213:173321. doi: 10.1016/j.pbb.2021.173321. Epub 2022 Jan 15. Pharmacol Biochem Behav. 2022. PMID: 35041859 Review.
-
The value of human epileptic tissue in the characterization and development of novel antiepileptic drugs: The example of CERC-611 and KRM-II-81.Brain Res. 2019 Nov 1;1722:146356. doi: 10.1016/j.brainres.2019.146356. Epub 2019 Jul 29. Brain Res. 2019. PMID: 31369732 Review.
Cited by
-
Design, synthesis and characterization of novel gamma‑aminobutyric acid type A receptor ligands.ARKIVOC. 2020;2020(Pt 7):242-256. doi: 10.24820/ark.5550190.p011.398. Epub 2020 Dec 2. ARKIVOC. 2020. PMID: 33642954 Free PMC article.
-
GABAkines - Advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors.Pharmacol Ther. 2022 Jun;234:108035. doi: 10.1016/j.pharmthera.2021.108035. Epub 2021 Nov 16. Pharmacol Ther. 2022. PMID: 34793859 Free PMC article. Review.
-
Identification of New Antiseizure Medication Candidates in Preclinical Animal Studies.Int J Mol Sci. 2023 Aug 24;24(17):13143. doi: 10.3390/ijms241713143. Int J Mol Sci. 2023. PMID: 37685950 Free PMC article. Review.
-
ENX-101, a GABAA receptor α2,3,5-selective positive allosteric modulator, displays antiseizure effects in rodent seizure and epilepsy models.Epilepsia. 2025 Jun;66(6):2124-2136. doi: 10.1111/epi.18340. Epub 2025 Mar 15. Epilepsia. 2025. PMID: 40088186 Free PMC article.
-
8-Substituted Triazolobenzodiazepines: In Vitro and In Vivo Pharmacology in Relation to Structural Docking at the α1 Subunit-Containing GABAA Receptor.Front Pharmacol. 2021 Apr 20;12:625233. doi: 10.3389/fphar.2021.625233. eCollection 2021. Front Pharmacol. 2021. PMID: 33959005 Free PMC article.
References
-
- Atack JR. (2011) GABAA receptor subtype-selective modulators. I. α2/α3-selective agonists as non-sedating anxiolytics. Curr Top Med Chem 11:1176–1202. - PubMed
-
- Barton ME, Klein BD, Wolf HH, White HS. (2001) Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res 47:217–227. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
