

Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis
- PMID: 31695033
- PMCID: PMC6834636
- DOI: 10.1038/s41467-019-13036-1
Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis
Abstract
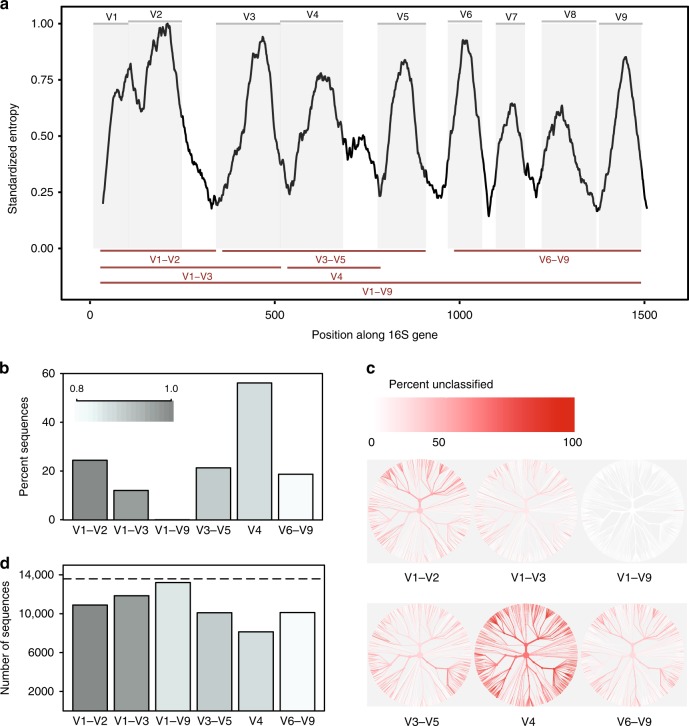
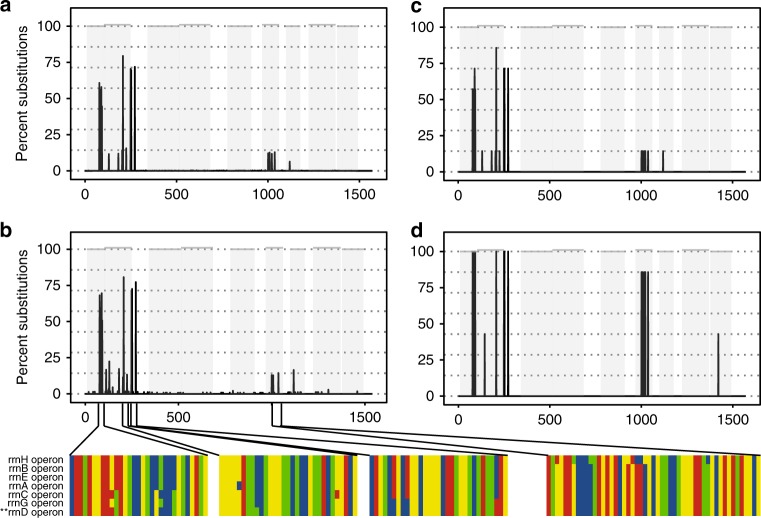
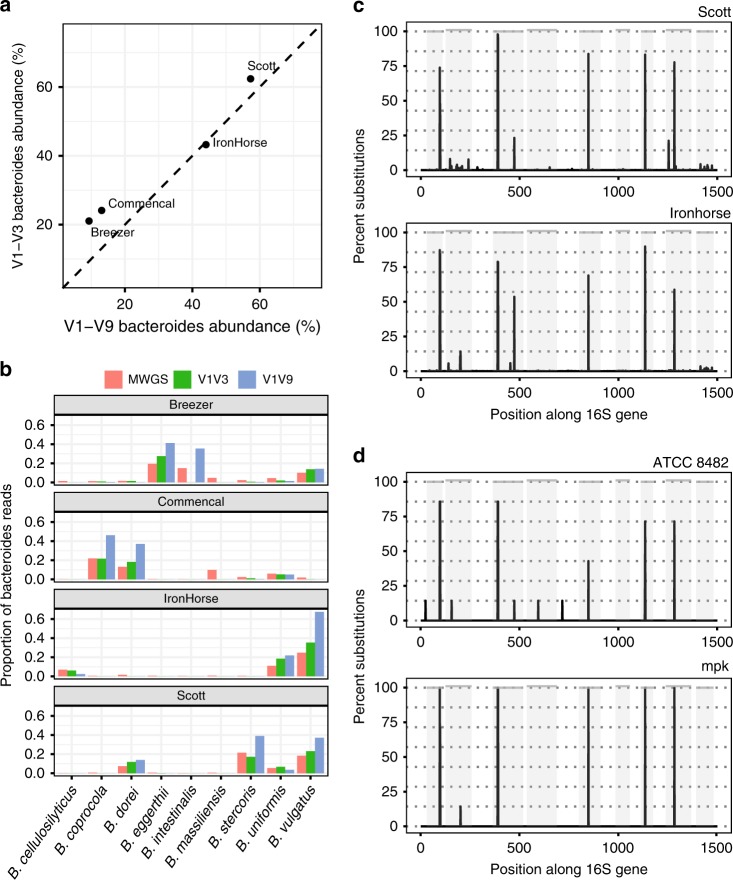
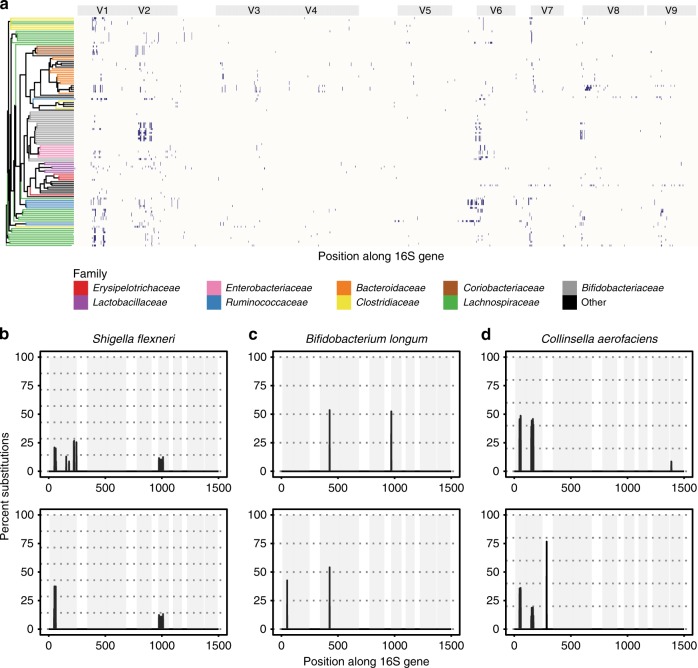
The 16S rRNA gene has been a mainstay of sequence-based bacterial analysis for decades. However, high-throughput sequencing of the full gene has only recently become a realistic prospect. Here, we use in silico and sequence-based experiments to critically re-evaluate the potential of the 16S gene to provide taxonomic resolution at species and strain level. We demonstrate that targeting of 16S variable regions with short-read sequencing platforms cannot achieve the taxonomic resolution afforded by sequencing the entire (~1500 bp) gene. We further demonstrate that full-length sequencing platforms are sufficiently accurate to resolve subtle nucleotide substitutions (but not insertions/deletions) that exist between intragenomic copies of the 16S gene. In consequence, we argue that modern analysis approaches must necessarily account for intragenomic variation between 16S gene copies. In particular, we demonstrate that appropriate treatment of full-length 16S intragenomic copy variants has the potential to provide taxonomic resolution of bacterial communities at species and strain level.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
