

Empirical design of a variant quality control pipeline for whole genome sequencing data using replicate discordance
- PMID: 31695094
- PMCID: PMC6834861
- DOI: 10.1038/s41598-019-52614-7
Empirical design of a variant quality control pipeline for whole genome sequencing data using replicate discordance
Abstract
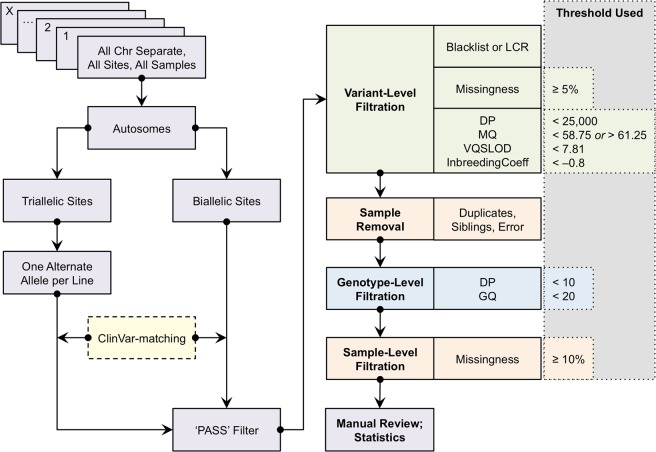
The success of next-generation sequencing depends on the accuracy of variant calls. Few objective protocols exist for QC following variant calling from whole genome sequencing (WGS) data. After applying QC filtering based on Genome Analysis Tool Kit (GATK) best practices, we used genotype discordance of eight samples that were sequenced twice each to evaluate the proportion of potentially inaccurate variant calls. We designed a QC pipeline involving hard filters to improve replicate genotype concordance, which indicates improved accuracy of genotype calls. Our pipeline analyzes the efficacy of each filtering step. We initially applied this strategy to well-characterized variants from the ClinVar database, and subsequently to the full WGS dataset. The genome-wide biallelic pipeline removed 82.11% of discordant and 14.89% of concordant genotypes, and improved the concordance rate from 98.53% to 99.69%. The variant-level read depth filter most improved the genome-wide biallelic concordance rate. We also adapted this pipeline for triallelic sites, given the increasing proportion of multiallelic sites as sample sizes increase. For triallelic sites containing only SNVs, the concordance rate improved from 97.68% to 99.80%. Our QC pipeline removes many potentially false positive calls that pass in GATK, and may inform future WGS studies prior to variant effect analysis.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Grants and funding
- R01 AG042188/AG/NIA NIH HHS/United States
- R01 AG046949/AG/NIA NIH HHS/United States
- R01 AG057909/AG/NIA NIH HHS/United States
- R01 AG 046949/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)/International
- R01 AG 042188/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)/International
- R01 AG 618381/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)/International
- K08 AG054727/AG/NIA NIH HHS/United States
- P01 AG021654/AG/NIA NIH HHS/United States
- P01 AG 021654/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)/International
- K08AG054727/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)/International
- P30 AG038072/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
