

An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome
- PMID: 31695190
- PMCID: PMC7231560
- DOI: 10.1038/s41594-019-0323-x
An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome
Abstract
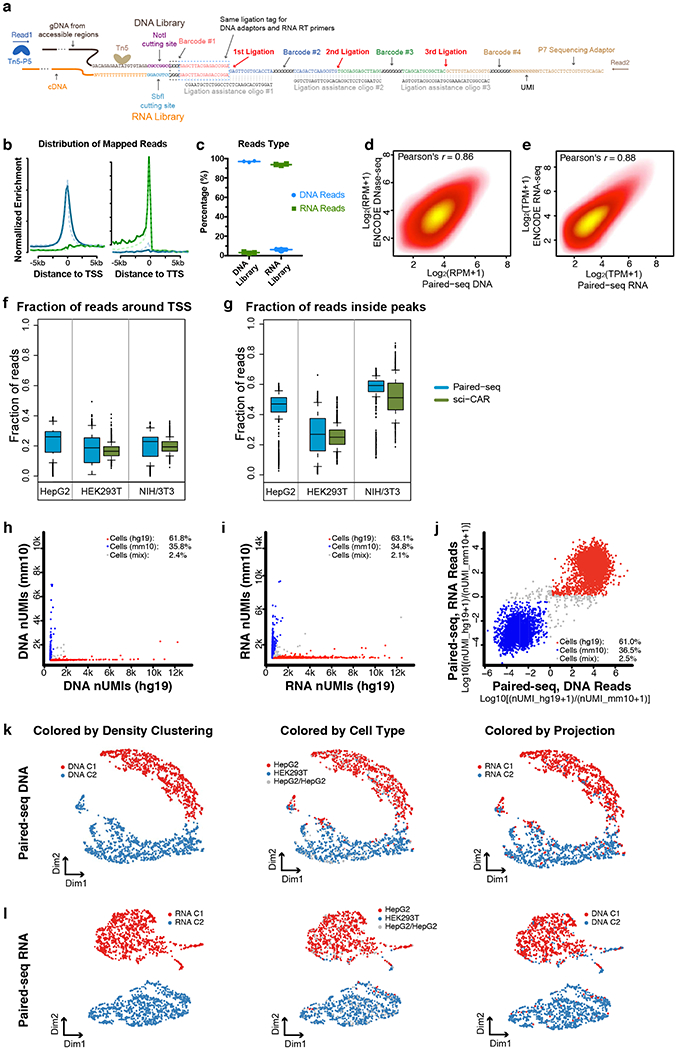
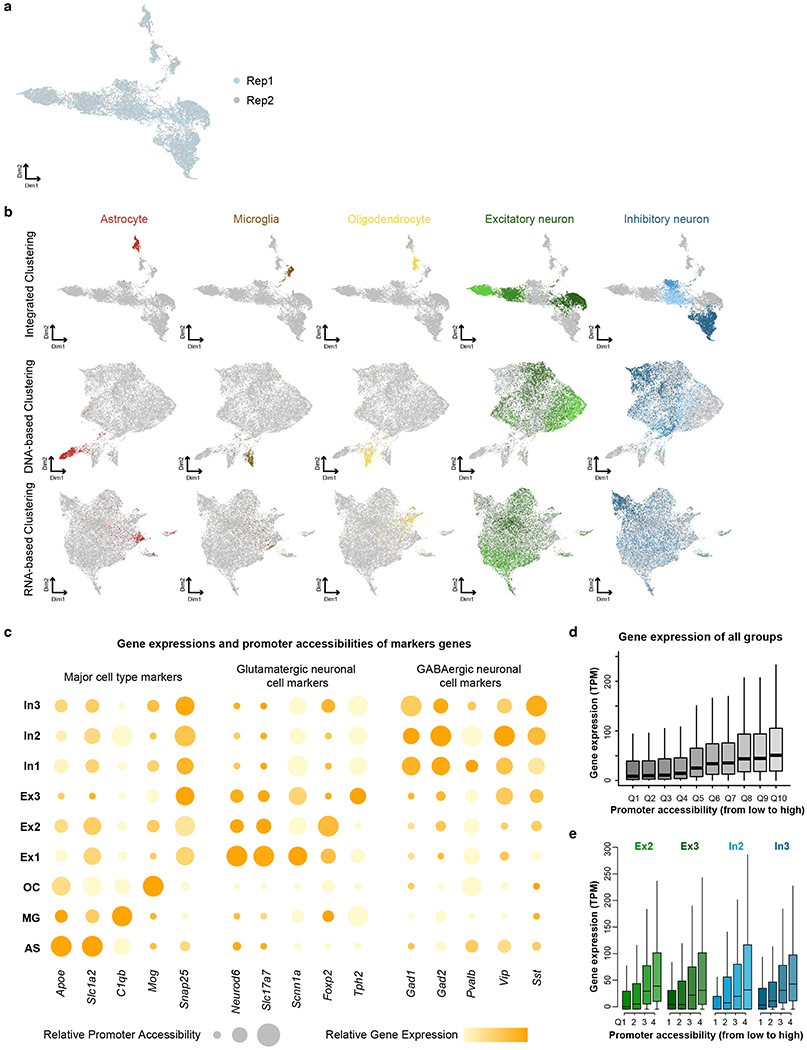
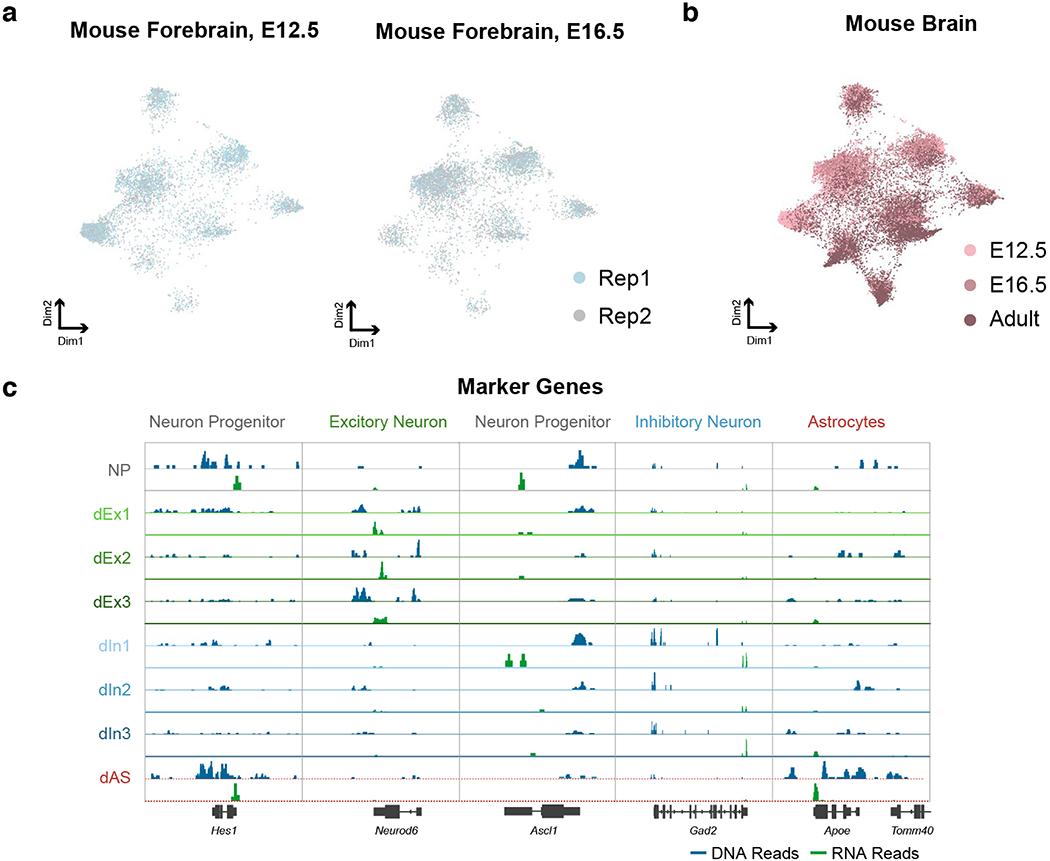
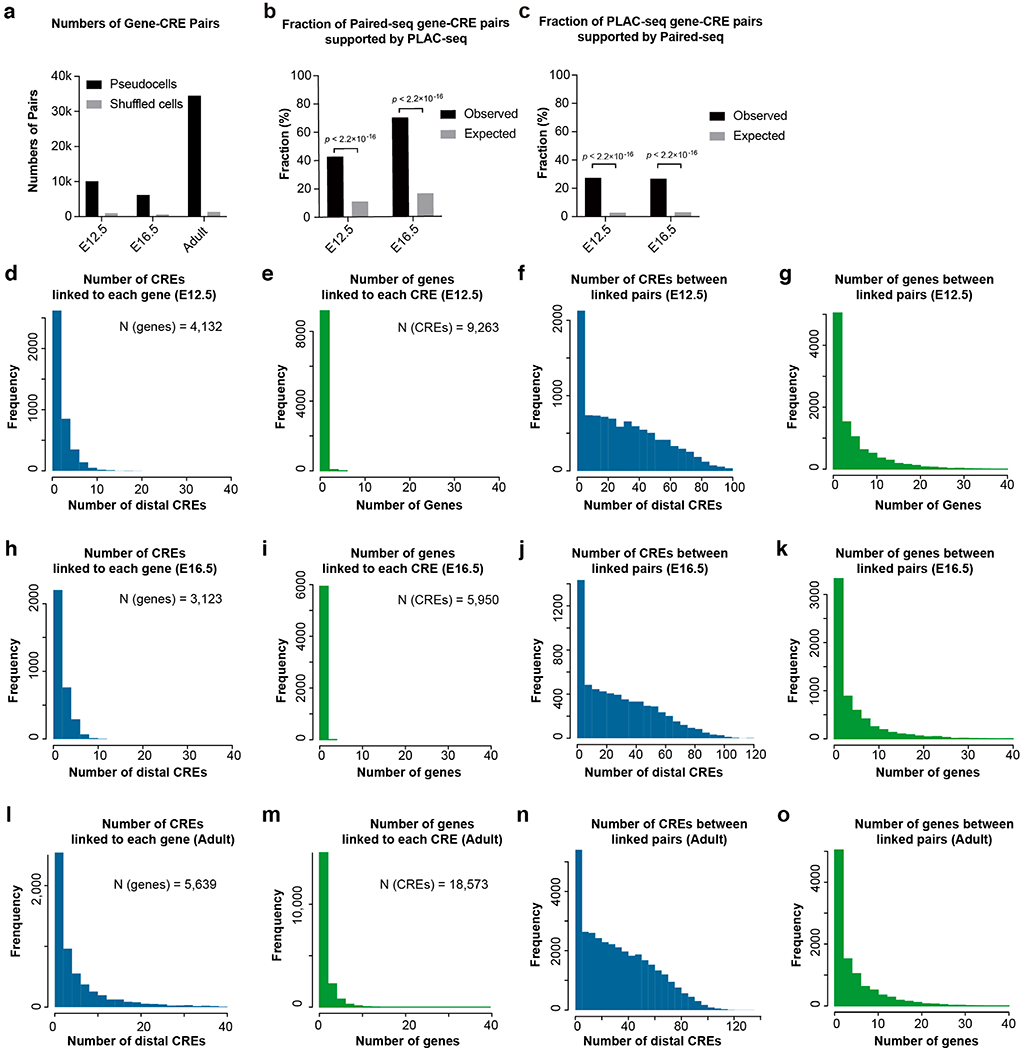
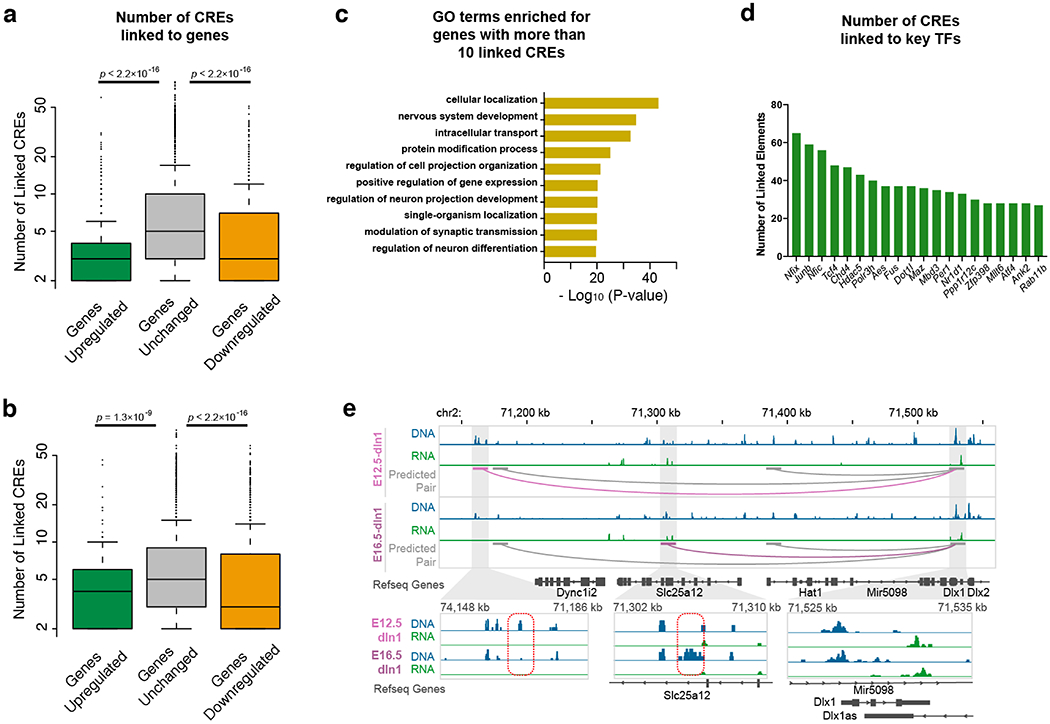
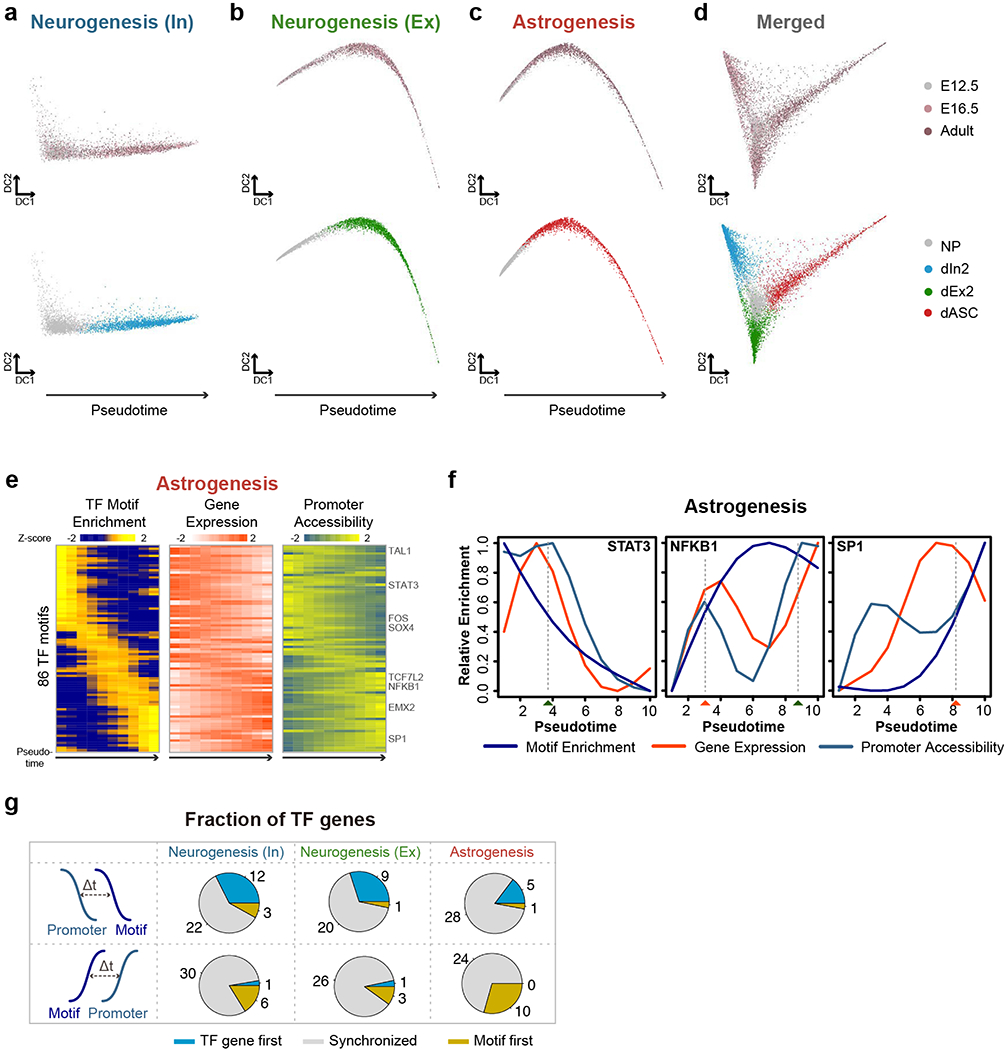
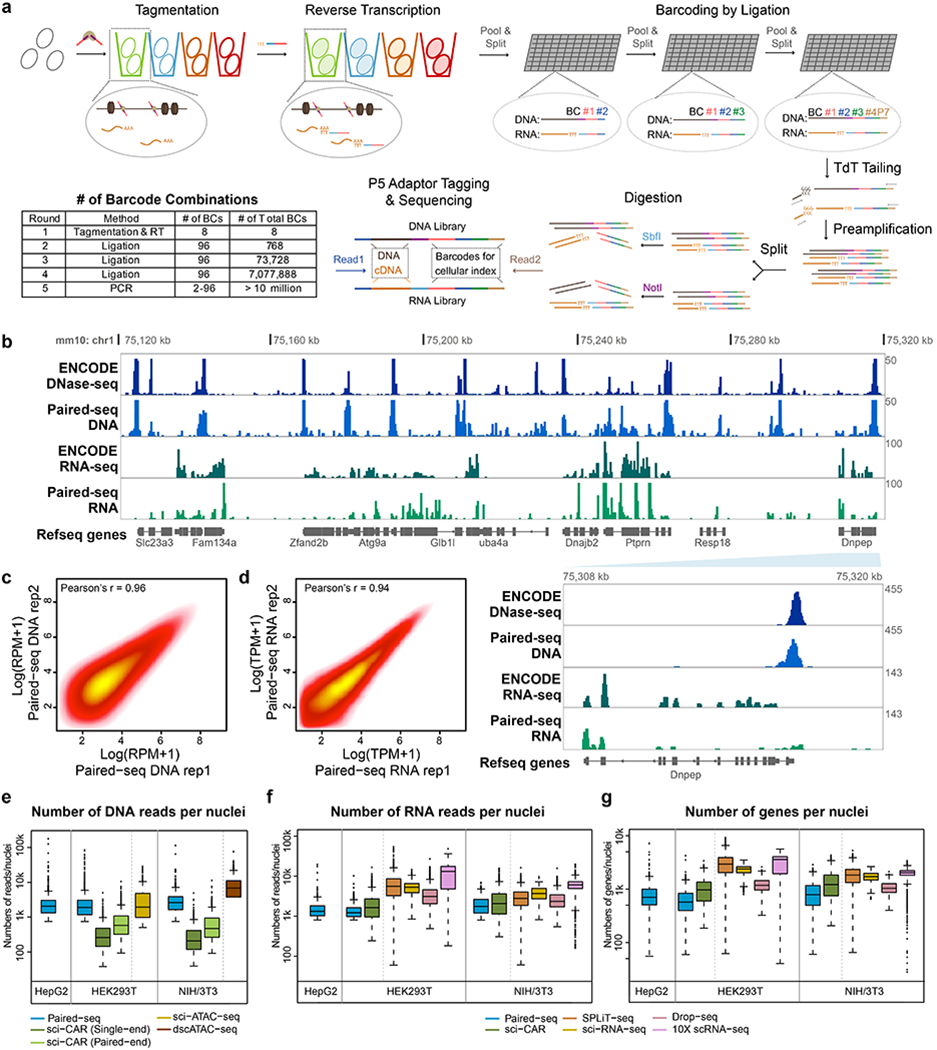
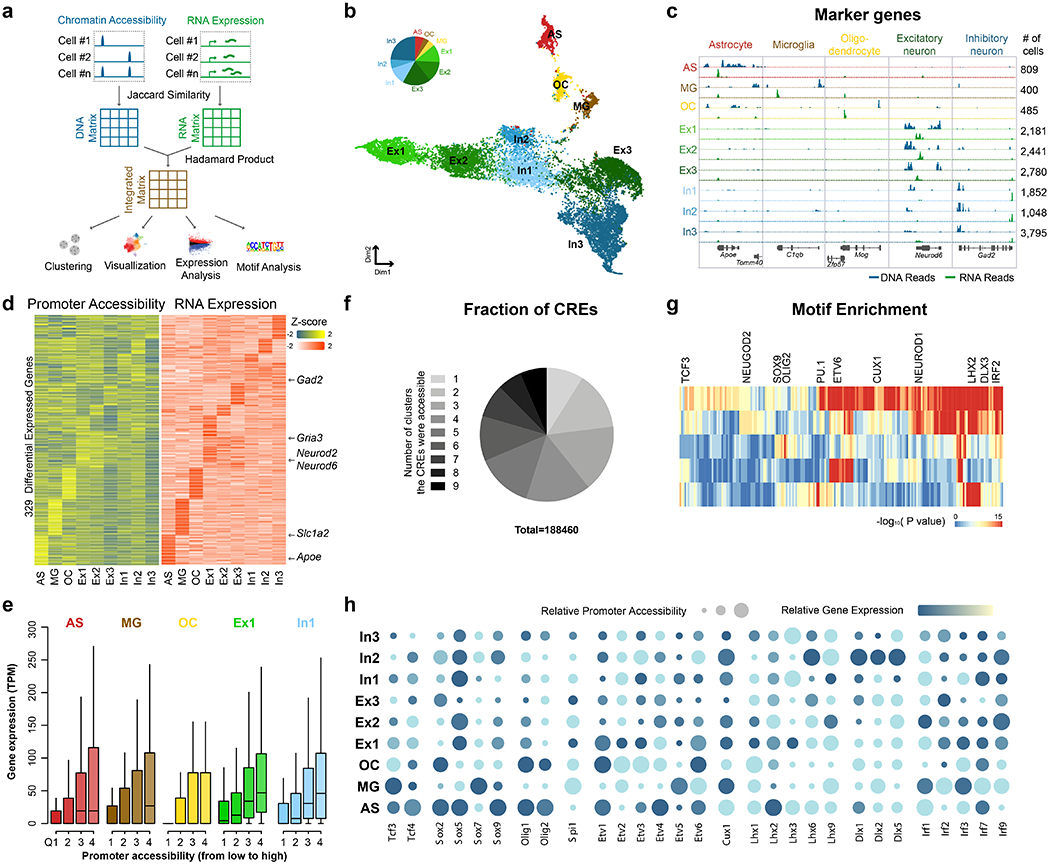
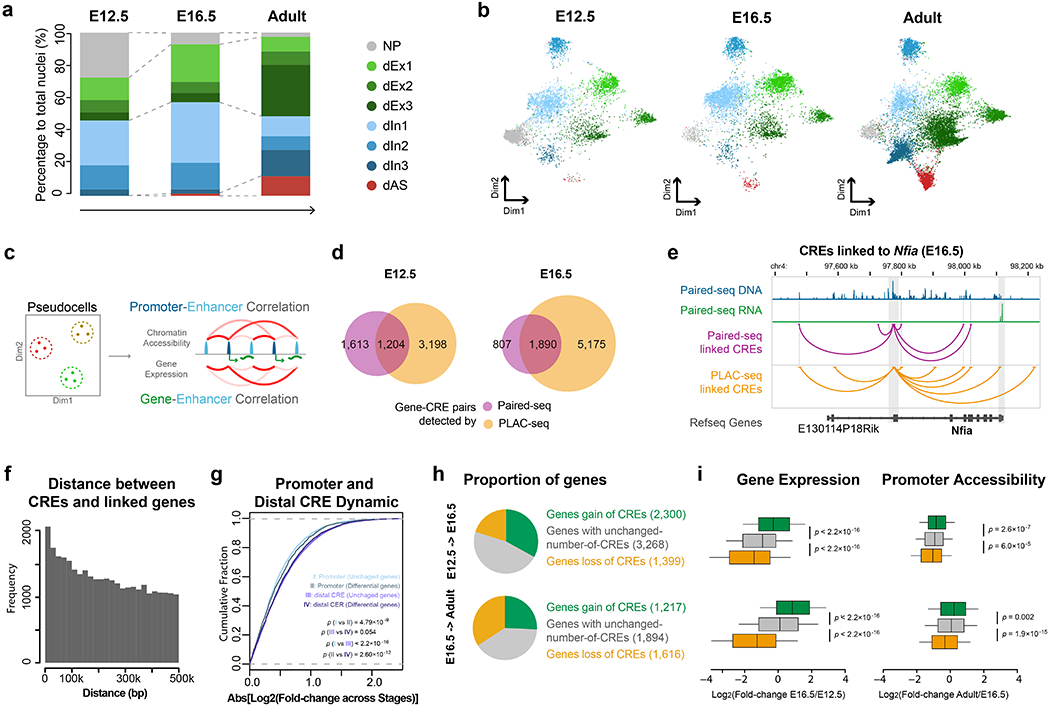
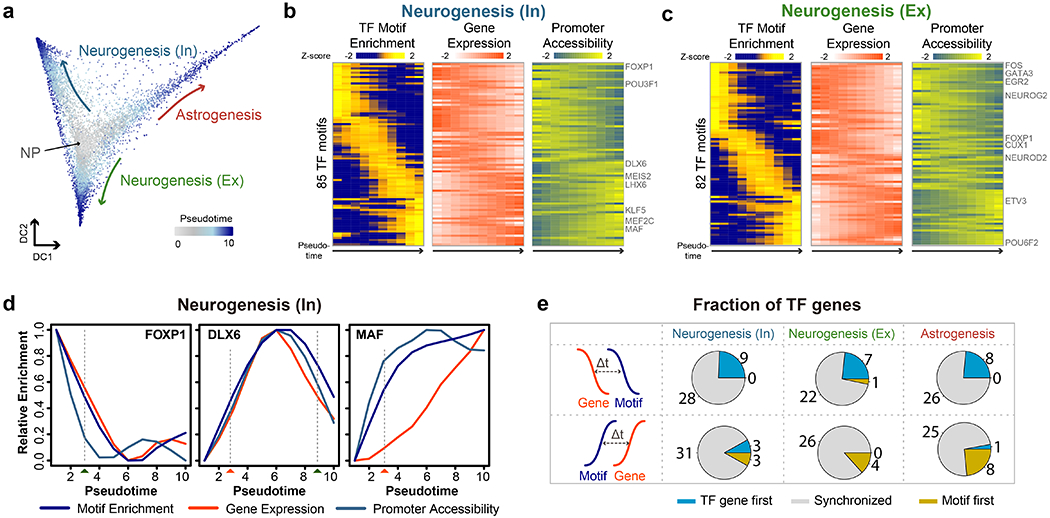
Simultaneous profiling of transcriptome and chromatin accessibility within single cells is a powerful approach to dissect gene regulatory programs in complex tissues. However, current tools are limited by modest throughput. We now describe an ultra high-throughput method, Paired-seq, for parallel analysis of transcriptome and accessible chromatin in millions of single cells. We demonstrate the utility of Paired-seq for analyzing the dynamic and cell-type-specific gene regulatory programs in complex tissues by applying it to mouse adult cerebral cortex and fetal forebrain. The joint profiles of a large number of single cells allowed us to deconvolute the transcriptome and open chromatin landscapes in the major cell types within these brain tissues, infer putative target genes of candidate enhancers, and reconstruct the trajectory of cellular lineages within the developing forebrain.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
