

Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson's disease
- PMID: 31703585
- PMCID: PMC6842240
- DOI: 10.1186/s13024-019-0339-z
Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson's disease
Erratum in
-
Correction to: Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson's disease.Mol Neurodegener. 2020 Jan 15;15(1):6. doi: 10.1186/s13024-019-0355-z. Mol Neurodegener. 2020. PMID: 31937358 Free PMC article.
Abstract
Background: Haploinsufficiency in the Gaucher disease GBA gene, which encodes the lysosomal glucocerebrosidase GBA, and ageing represent major risk factors for developing Parkinson's disease (PD). Recently, more than fifty other lysosomal storage disorder gene variants have been identified in PD, implicating lysosomal dysfunction more broadly as a key risk factor for PD. Despite the evidence of multiple lysosomal genetic risks, it remains unclear how sphingolipid hydrolase activities, other than GBA, are altered with ageing or in PD. Moreover, it is not fully known if levels of glycosphingolipid substrates for these enzymes change in vulnerable brain regions of PD. Finally, little is known about the levels of complex gangliosides in substantia nigra which may play a significant role in ageing and PD.
Methods: To study sphingolipid hydrolase activities and glycosphingolipid expression in ageing and in PD, two independent cohorts of human substantia nigra tissues were obtained. Fluorescent 4-methylumbelliferone assays were used to determine multiple enzyme activities. The lysosomal GBA and non-lysosomal GBA2 activities were distinguished using the inhibitor NB-DGJ. Sensitive and quantitative normal-phase HPLC was performed to study glycosphingolipid levels. In addition, glycosphingolipid levels in cerebrospinal fluid and serum were analysed as possible biomarkers for PD.
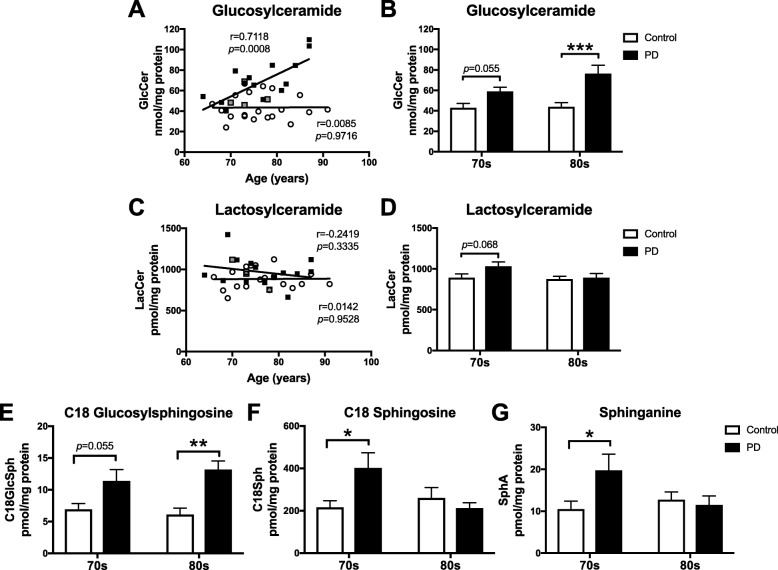
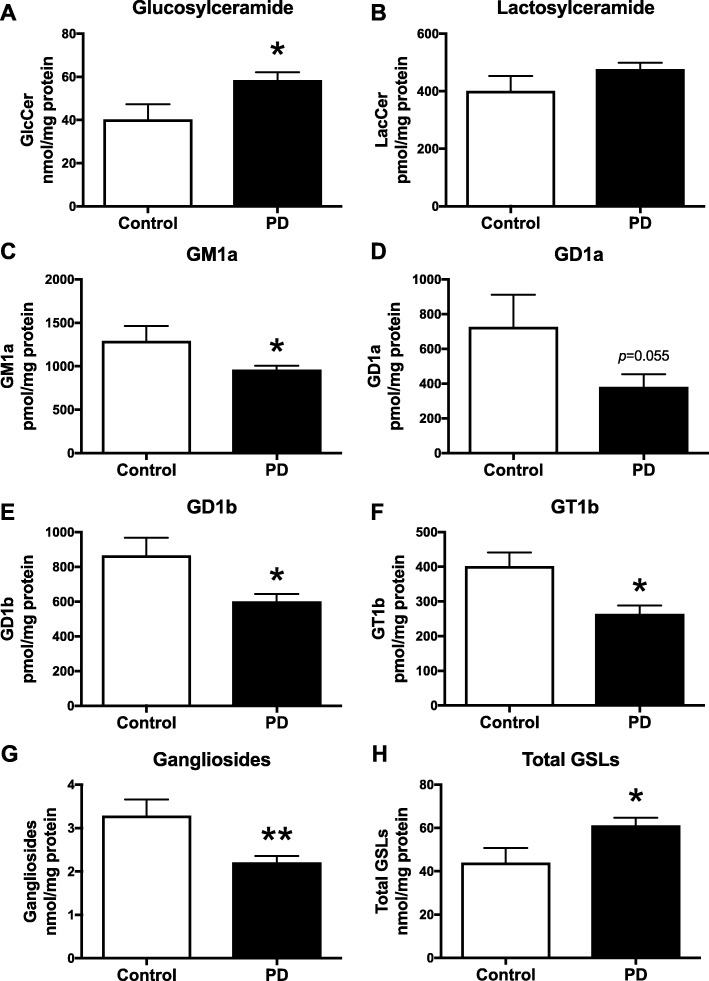
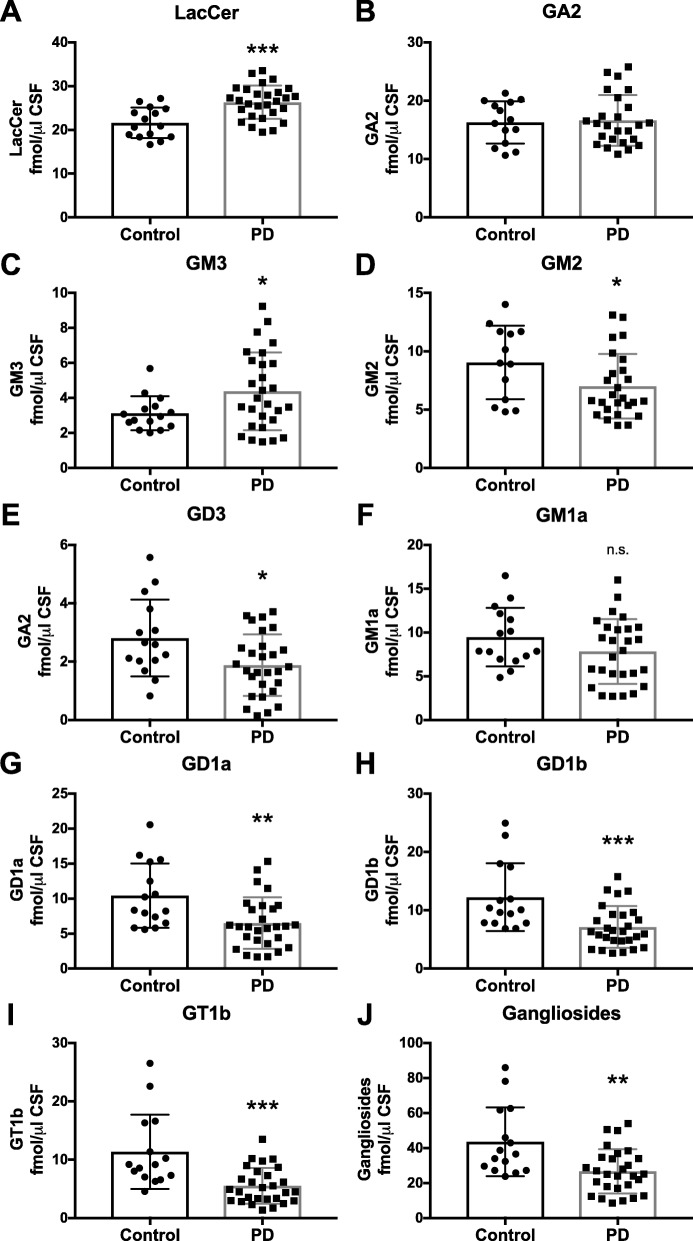
Results: The present study demonstrates, in two independent cohorts of human post-mortem substantia nigra, that sporadic PD is associated with deficiencies in multiple lysosomal hydrolases (e.g. α-galactosidase and β-hexosaminidase), in addition to reduced GBA and GBA2 activities and concomitant glycosphingolipid substrate accumulation. Furthermore, the data show significant reductions in levels of complex gangliosides (e.g. GM1a) in substantia nigra, CSF and serum in ageing, PD, and REM sleep behaviour disorder, which is a strong predictor of PD.
Conclusions: These findings conclusively demonstrate reductions in GBA activity in the parkinsonian midbrain, and for the first time, reductions in the activity of several other sphingolipid hydrolases. Furthermore, significant reductions were seen in complex gangliosides in PD and ageing. The diminished activities of these lysosomal hydrolases, the glycosphingolipid substrate accumulation, and the reduced levels of complex gangliosides are likely major contributors to the primary development of the pathology seen in PD and related disorders with age.
Keywords: Ageing; Ganglioside; Glucocerebrosidase; Glycosphingolipid; Lysosome; Neurodegeneration; Parkinson’s disease.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
