

Advances in Treatment of Spinal Muscular Atrophy - New Phenotypes, New Challenges, New Implications for Care
- PMID: 31707373
- PMCID: PMC7029319
- DOI: 10.3233/JND-190424
Advances in Treatment of Spinal Muscular Atrophy - New Phenotypes, New Challenges, New Implications for Care
Abstract
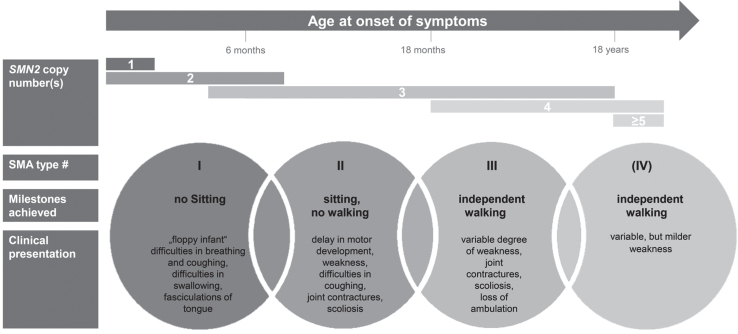
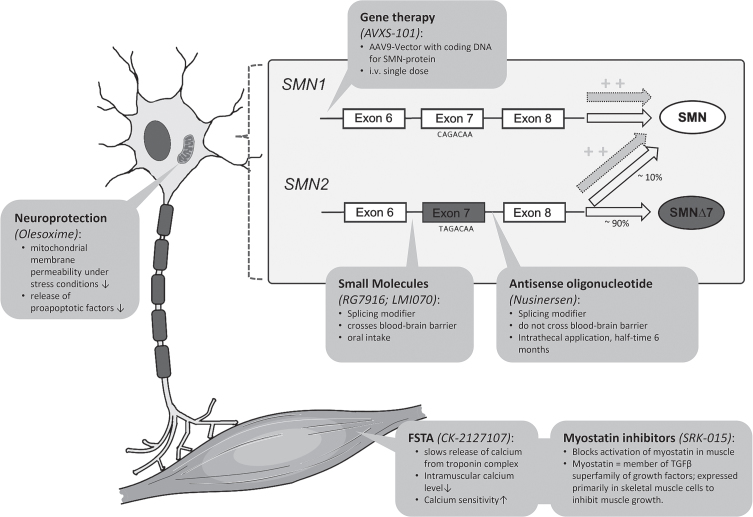
Spinal Muscular Atrophy (SMA) is caused by autosomal recessive mutations in SMN1 and results in the loss of motor neurons and progressive muscle weakness. The spectrum of disease severity ranges from early onset with respiratory failure during the first months of life to a mild, adult-onset type with slow rate of progression. Over the past decade, new treatment options such as splicing modulation of SMN2 and SMN1 gene replacement by gene therapy have been developed. First drugs have been approved for treatment of patients with SMA and if initiated early they can significantly modify the natural course of the disease. As a consequence, newborn screening for SMA is explored and implemented in an increasing number of countries. However, available evidence for these new treatments is often limited to a small spectrum of patients concerning age and disease stage. In this review we provide an overview of available and emerging therapies for spinal muscular atrophy and we discuss new phenotypes and associated challenges in clinical care. Collection of real-world data with standardized outcome measures will be essential to improve both the understanding of treatment effects in patients of all SMA subtypes and the basis for clinical decision-making in SMA.
Keywords: Spinal muscular atrophy; antisense oligonucleotides; gene therapy; neonatal screening; outcome assessment; registries.
Conflict of interest statement
Dr. David C. Schorling, participated in workshops sponsored by Biogen and Roche.
Dr. Astrid Pechmann, received compensation for presentations and training activities from Biogen, received research funding from Biogen.
Prof. Dr. Janbernd Kirschner, received research funding and/or compensation for presentations and consulting services from Avexis, Biogen, Ionis Pharmaceuticals, Novartis, and Roche.
Figures
References
-
- Werdnig G. Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis. 1891. Arch Neurol. 1971;25:276–8. - PubMed
-
- Hoffmann J. Ueber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis. Dtsch Z Für Nervenheilkd. 1893;3:427–70. doi: 10.1007/BF01668496 - DOI
-
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–65. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
