

DNA double-strand breaks: a potential therapeutic target for neurodegenerative diseases
- PMID: 31707536
- PMCID: PMC7934912
- DOI: 10.1007/s10577-019-09617-x
DNA double-strand breaks: a potential therapeutic target for neurodegenerative diseases
Abstract
The complexity of neurodegeneration restricts the ability to understand and treat the neurological disorders affecting millions of people worldwide. Therefore, there is an unmet need to develop new and more effective therapeutic strategies to combat these devastating conditions and that will only be achieved with a better understanding of the biological mechanism associated with disease conditions. Recent studies highlight the role of DNA damage, particularly, DNA double-strand breaks (DSBs), in the progression of neuronal loss in a broad spectrum of human neurodegenerative diseases. This is not unexpected because neurons are prone to DNA damage due to their non-proliferative nature and high metabolic activity. However, it is not clear if DSBs is a primary driver of neuronal loss in disease conditions or simply occurs concomitant with disease progression. Here, we provide evidence that supports a critical role of DSBs in the pathogenesis of the neurodegenerative diseases. Among different kinds of DNA damages, DSBs are the most harmful and perilous type of DNA damage and can lead to cell death if left unrepaired or repaired with error. In this review, we explore the current state of knowledge regarding the role of DSBs repair mechanisms in preserving neuronal function and survival and describe how DSBs could drive the molecular mechanisms resulting in neuronal death in neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. We also discuss the potential implications of DSBs as a novel therapeutic target and prognostic marker in patients with neurodegenerative conditions.
Keywords: Alzheimer’s disease; DNA damage; DNA repair; Genomic instability, Neurodegeneration; Parkinson’s disease.
Conflict of interest statement
Figures
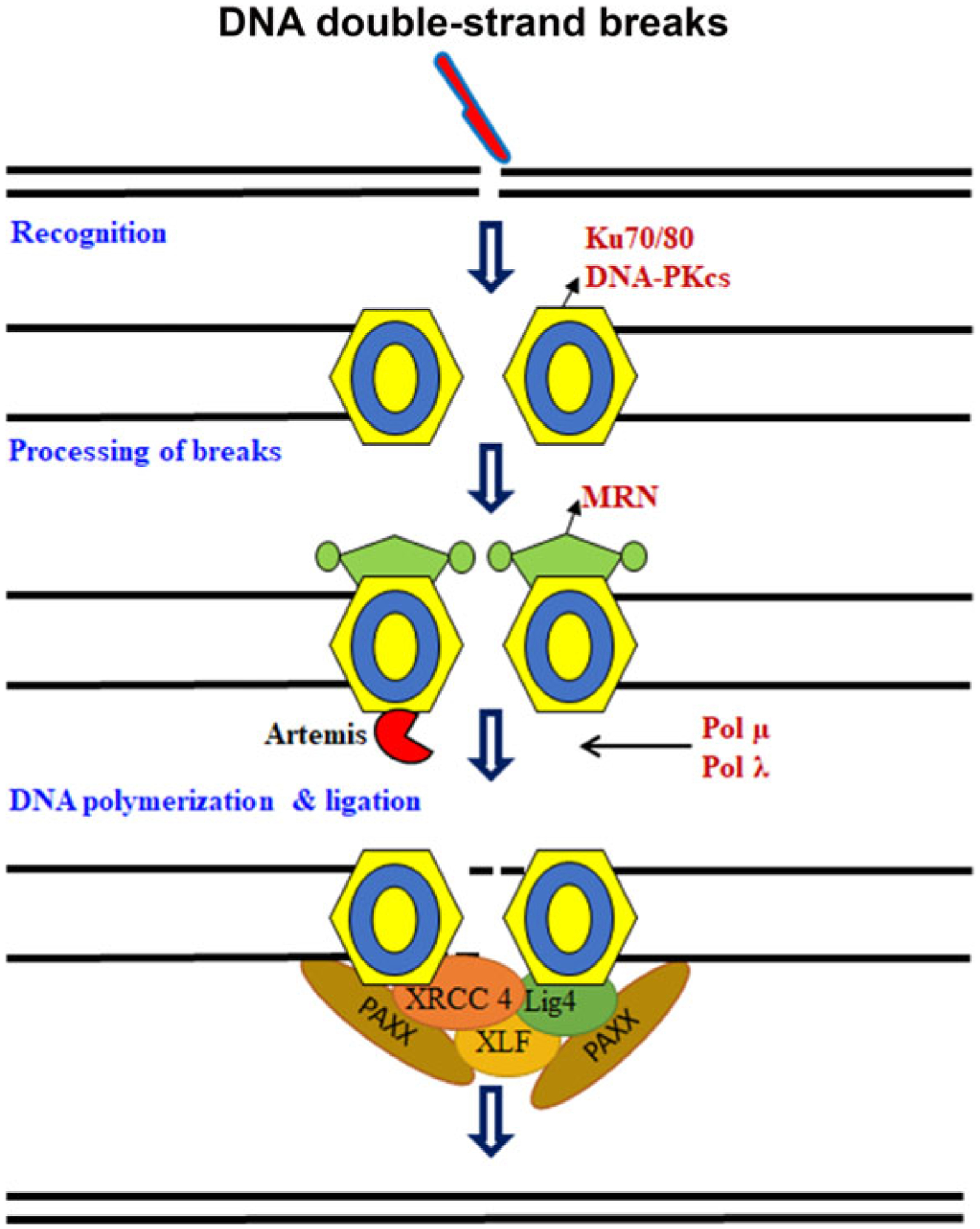
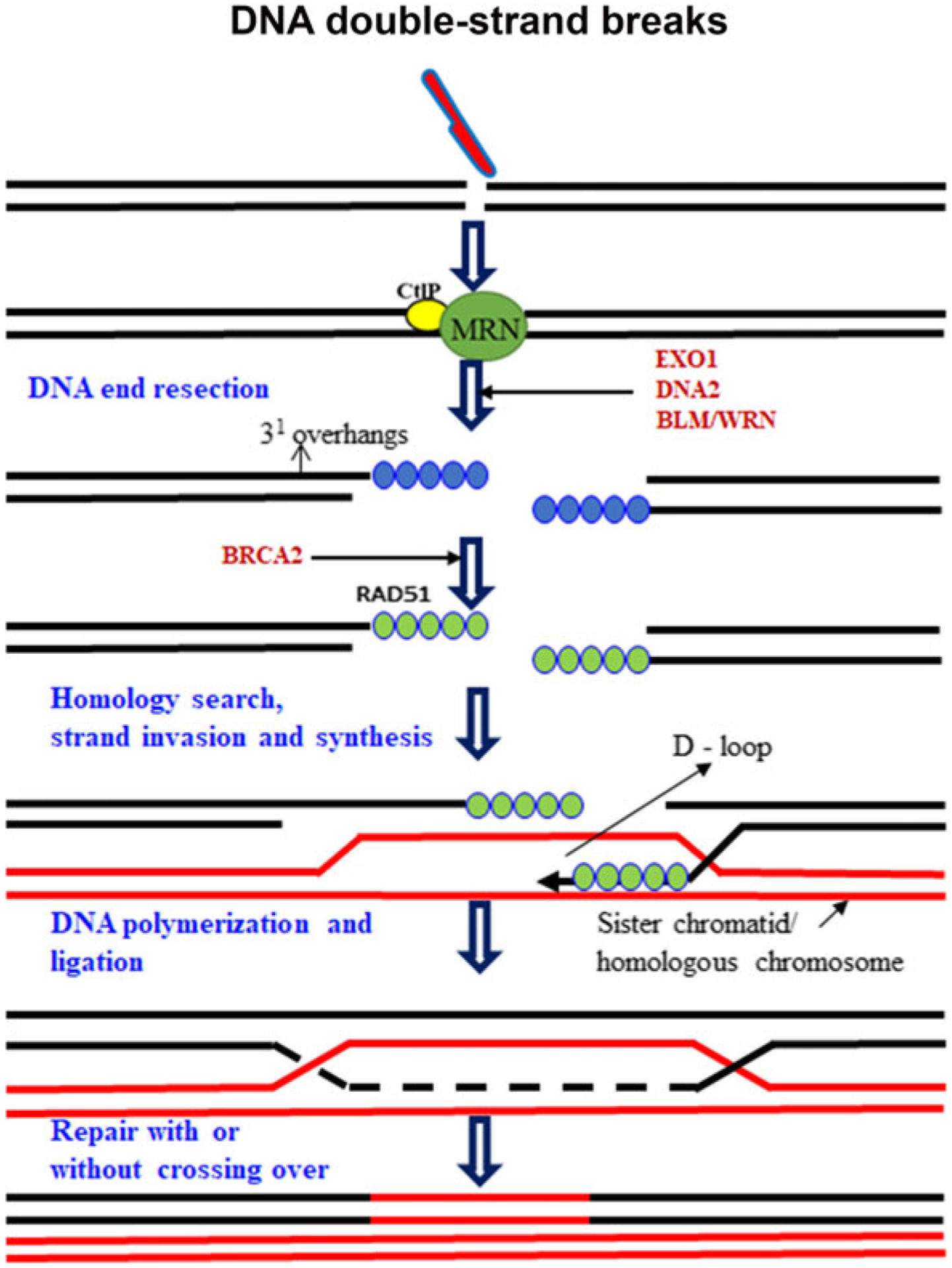
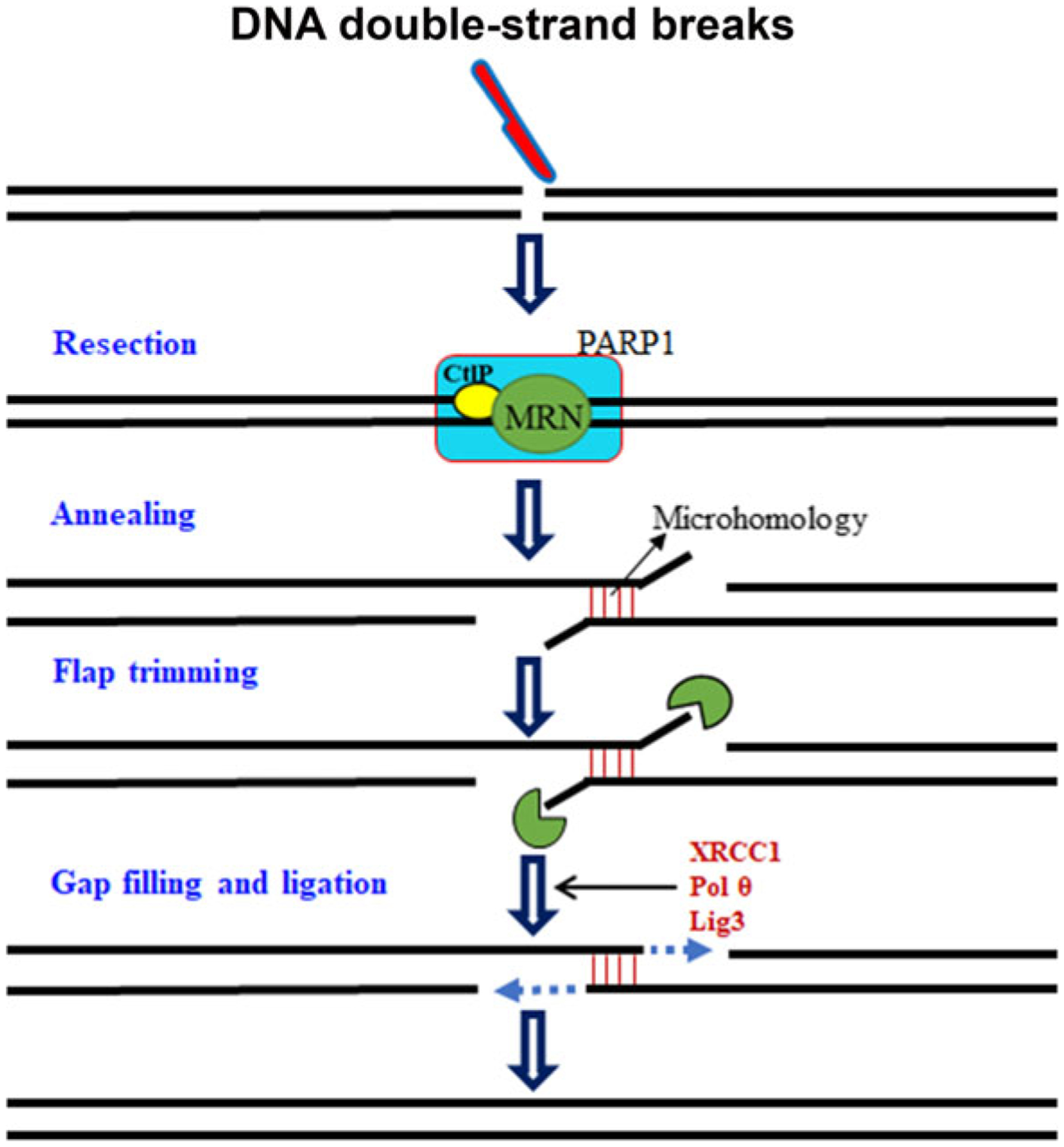
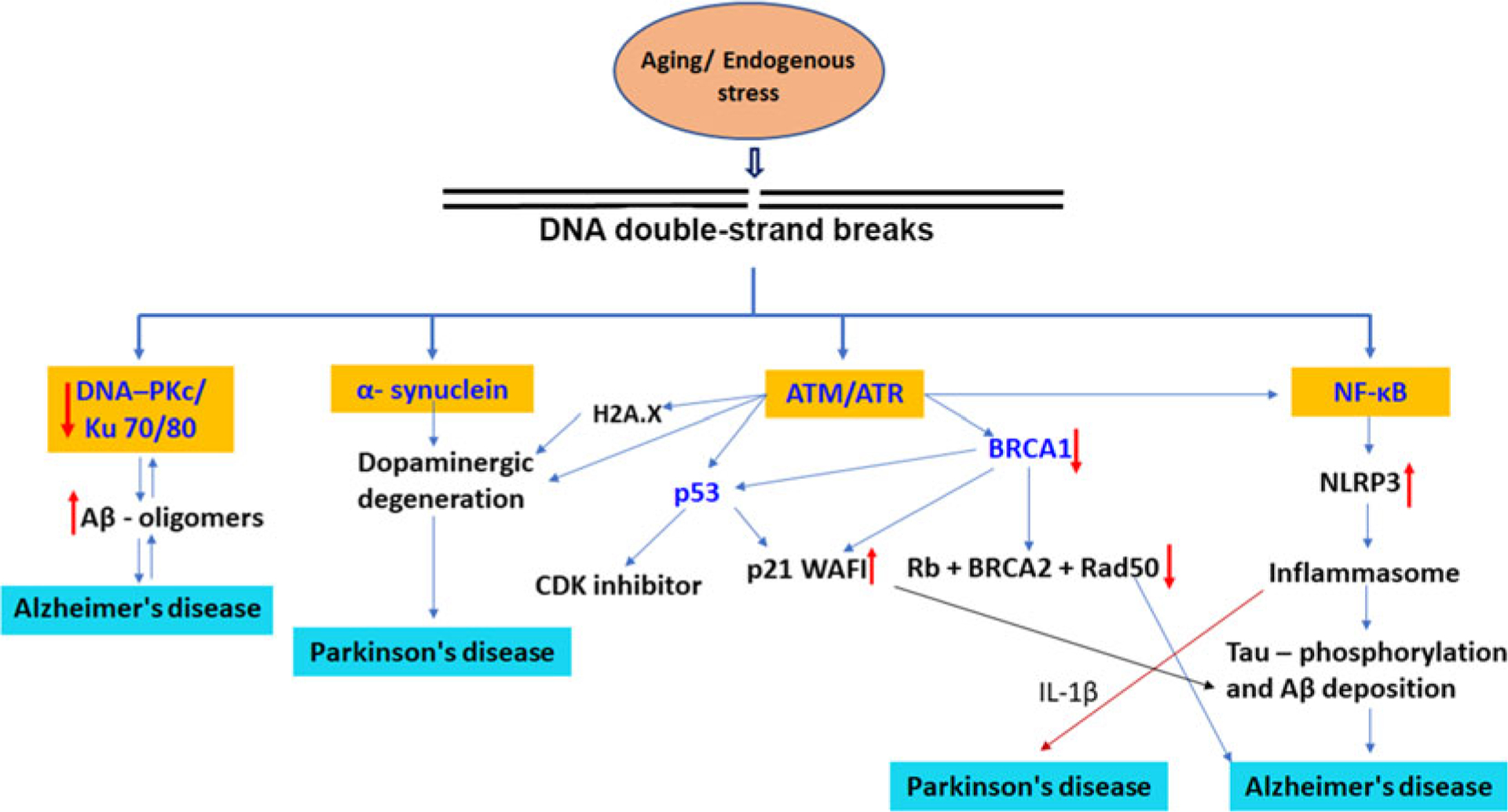
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
