

SNARE proteins rescue impaired autophagic flux in Down syndrome
- PMID: 31714914
- PMCID: PMC6850524
- DOI: 10.1371/journal.pone.0223254
SNARE proteins rescue impaired autophagic flux in Down syndrome
Abstract
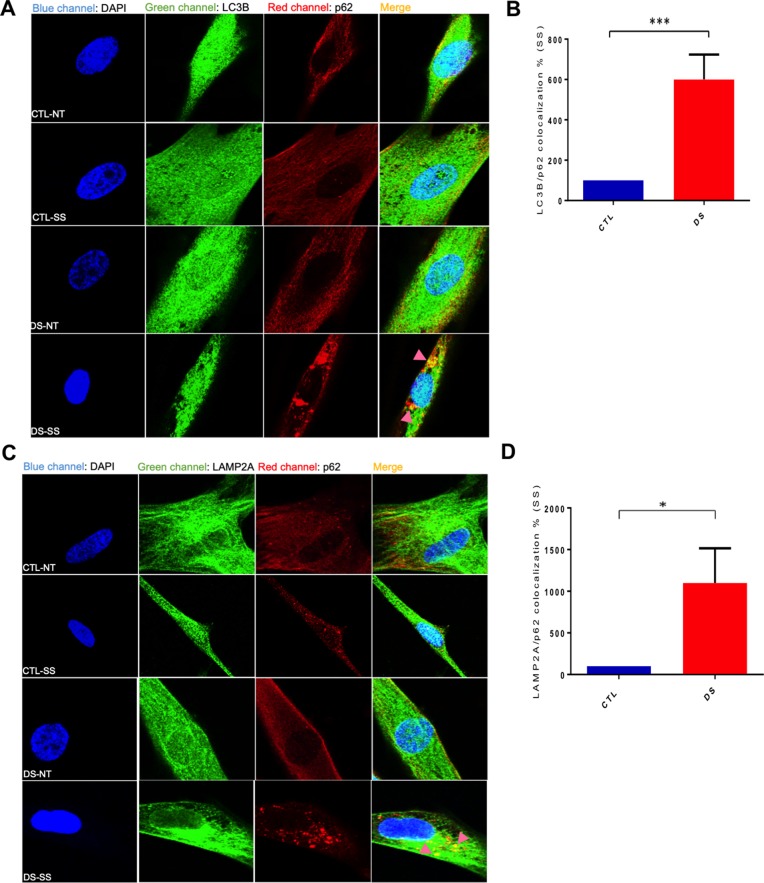
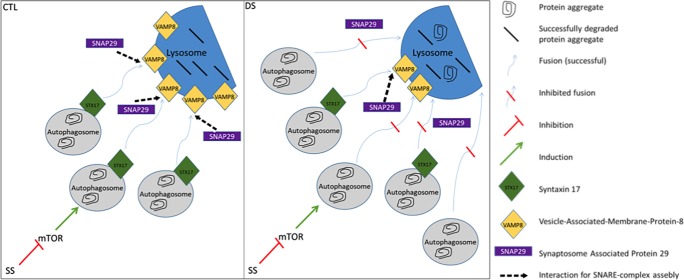
Down syndrome (DS) is a chromosomal disorder caused by trisomy of chromosome 21 (Ts21). Unbalanced karyotypes can lead to dysfunction of the proteostasis network (PN) and disrupted proteostasis is mechanistically associated with multiple DS comorbidities. Autophagy is a critical component of the PN that has not previously been investigated in DS. Based on our previous observations of PN disruption in DS, we investigated possible dysfunction of the autophagic machinery in human DS fibroblasts and other DS cell models. Following induction of autophagy by serum starvation, DS fibroblasts displayed impaired autophagic flux indicated by autophagolysosome accumulation and elevated p62, NBR1, and LC3-II abundance, compared to age- and sex-matched, euploid (CTL) fibroblasts. While lysosomal physiology was unaffected in both groups after serum starvation, we observed decreased basal abundance of the Soluble N-ethylmaleimide-sensitive-factor Attachment protein Receptor (SNARE) family members syntaxin 17 (STX17) and Vesicle Associated Membrane Protein 8 (VAMP8) indicating that decreased autophagic flux in DS is due at least in part to a possible impairment of autophagosome-lysosome fusion. This conclusion was further supported by the observation that over-expression of either STX17 or VAMP8 in DS fibroblasts restored autophagic degradation and reversed p62 accumulation. Collectively, our results indicate that impaired autophagic clearance is a characteristic of DS cells that can be reversed by enhancement of SNARE protein expression and provides further evidence that PN disruption represents a candidate mechanism for multiple aspects of pathogenesis in DS and a possible future target for therapeutic intervention.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
