

Mucus, mucins, and cystic fibrosis
- PMID: 31715083
- PMCID: PMC6853602
- DOI: 10.1002/ppul.24530
Mucus, mucins, and cystic fibrosis
Abstract
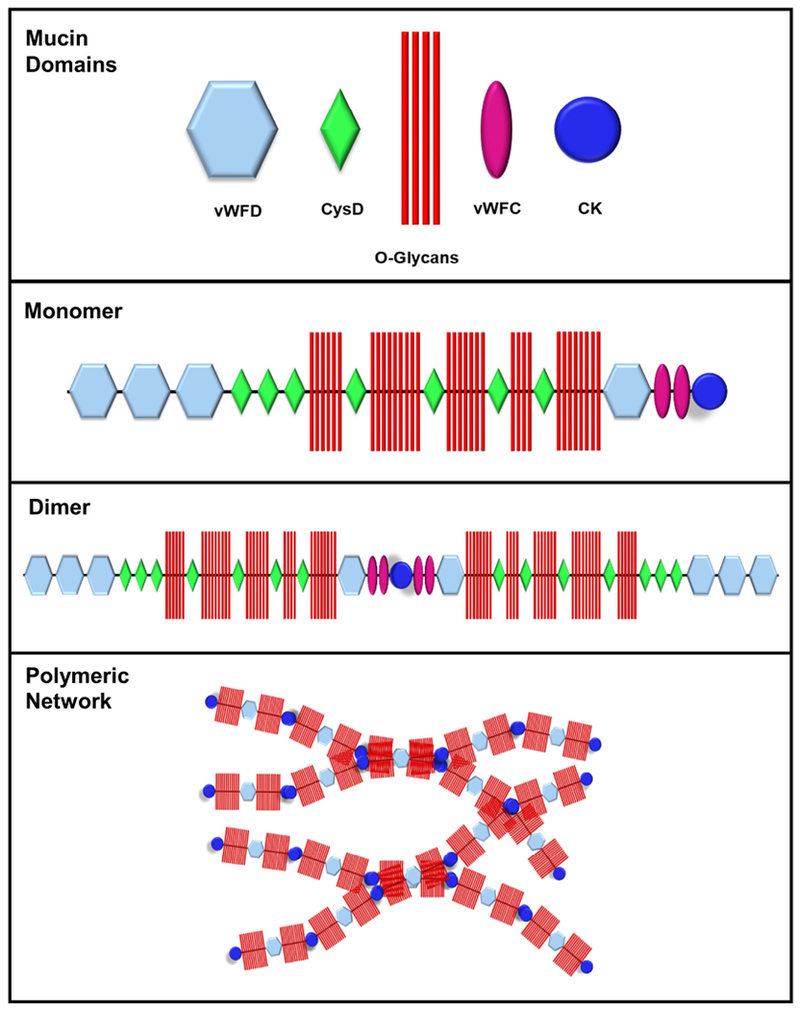
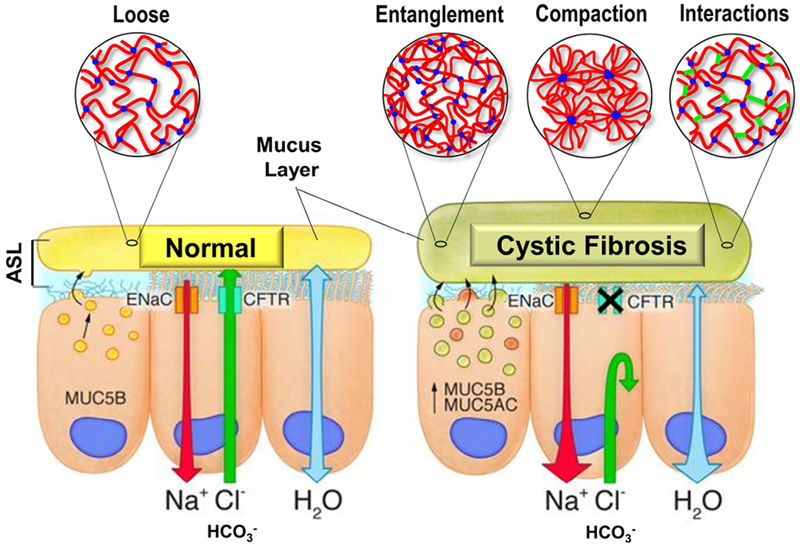
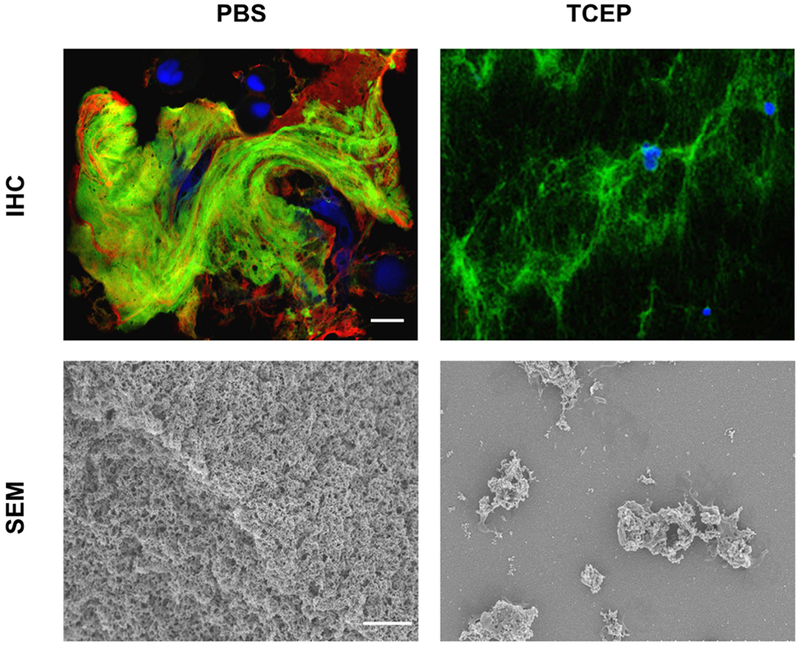
Cystic fibrosis (CF) is both the most common and most lethal genetic disease in the Caucasian population. CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and is characterized by the accumulation of thick, adherent mucus plaques in multiple organs, of which the lungs, gastrointestinal tract and pancreatic ducts are the most commonly affected. A similar pathogenesis cascade is observed in all of these organs: loss of CFTR function leads to altered ion transport, consisting of decreased chloride and bicarbonate secretion via the CFTR channel and increased sodium absorption via epithelial sodium channel upregulation. Mucosa exposed to changes in ionic concentrations sustain severe pathophysiological consequences. Altered mucus biophysical properties and weakened innate defense mechanisms ensue, furthering the progression of the disease. Mucins, the high-molecular-weight glycoproteins responsible for the viscoelastic properties of the mucus, play a key role in the disease but the actual mechanism of mucus accumulation is still undetermined. Multiple hypotheses regarding the impact of CFTR malfunction on mucus have been proposed and are reviewed here. (a) Dehydration increases mucin monomer entanglement, (b) defective Ca2+ chelation compromises mucin expansion, (c) ionic changes alter mucin interactions, and (d) reactive oxygen species increase mucin crosslinking. Although one biochemical change may dominate, it is likely that all of these mechanisms play some role in the progression of CF disease. This article discusses recent findings on the initial cause(s) of aberrant mucus properties in CF and examines therapeutic approaches aimed at correcting mucus properties.
Keywords: biochemical interactions; cftr; mucins; mucus; polymeric network; viscoelastic properties.
© 2019 Wiley Periodicals, Inc.
Conflict of interest statement
The authors have no conflict to disclose
Figures
References
-
- Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med. 2007;13(6):231–240. - PubMed
-
- Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008;372(9636):415–417. - PubMed
-
- Widdicombe JH, Wine JJ. Airway Gland Structure and Function. Physiol Rev. 2015;95(4):1241–1319. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
