

Cystic fibrosis precision therapeutics: Emerging considerations
- PMID: 31715091
- PMCID: PMC6871648
- DOI: 10.1002/ppul.24547
Cystic fibrosis precision therapeutics: Emerging considerations
Abstract
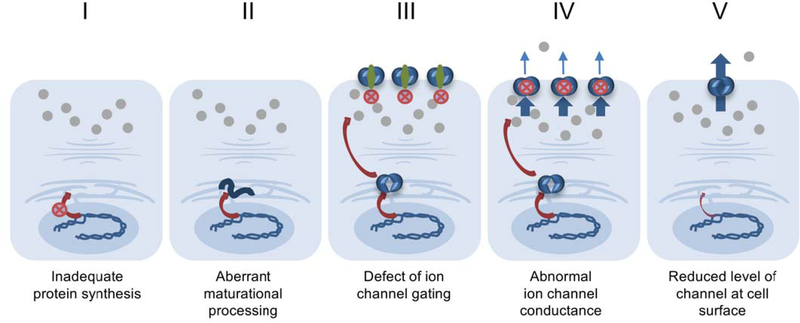
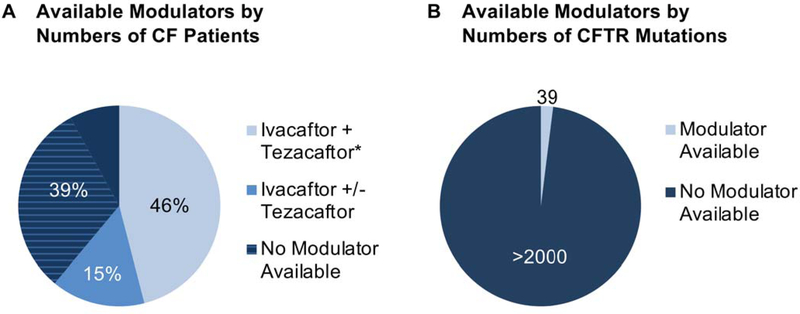
Small molecules that address fundamental defects underlying cystic fibrosis (CF), including modulators such as the approved drugs ivacaftor, lumacaftor, tezacaftor, and elexacaftor, have advanced dramatically over the past few years and are transforming care and prognosis among individuals with this disease. The new treatment strategies are predicated on established scientific insight concerning pathogenesis, and applying "personalized" or "precision" interventions for specific abnormalities of the cystic fibrosis transmembrane conductance regulator (CFTR). Even with the advent of highly effective triple drug combinations-which hold great promise for the majority of patients with CF worldwide-barriers to precision therapy remain. These include refractory CFTR variants (premature truncation codons, splice defects, large indels, severe missense mutations, and others) not addressed by available modulators, and access to leading-edge therapeutic compounds for patients with ultrarare forms of CF. In addition to describing the remarkable progress that has occurred regarding CF precision medicine, this review outlines some of the remaining challenges. The CF experience is emblematic of many conditions for which personalized interventions are actively being sought.
Keywords: combination drug therapy; investigational drugs; personalized medicine.
© 2019 Wiley Periodicals, Inc.
Figures
References
-
- Sorscher EJ. Cystic Fibrosis In: Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J, editors. Harrison’s principles of internal medicine. 20th edition New York: McGraw-Hill Education; 2018. Chapter 285,2 volumes (xli, 3528, I-214 pages).
-
- Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015; 373:1783–1784. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
